Downloaded 219 times




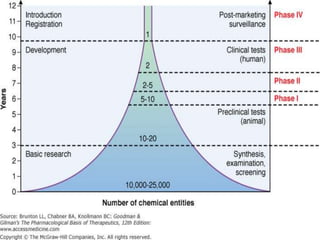


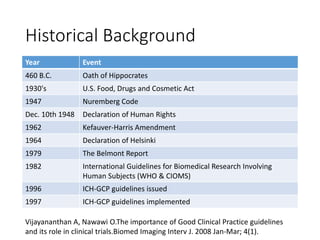






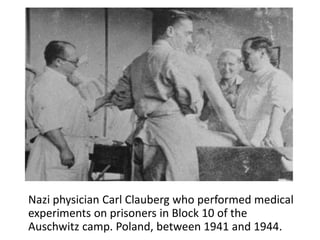

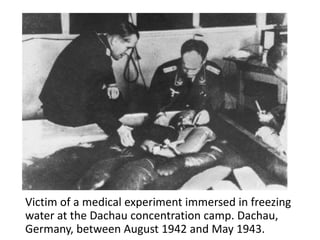
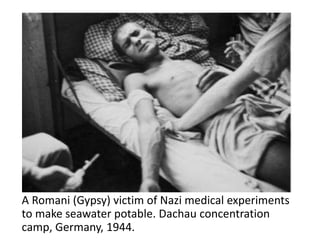
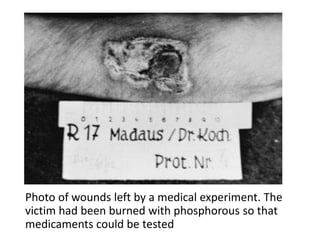











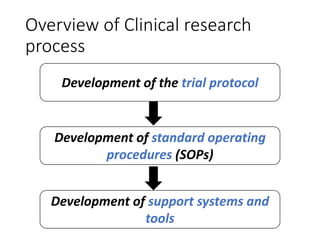

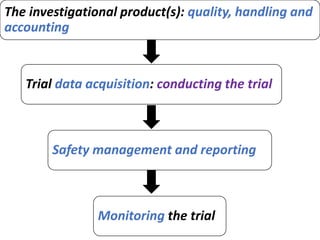
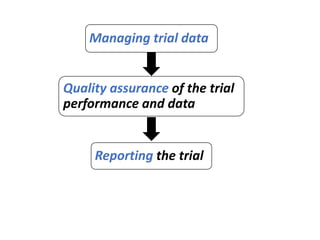



























This document provides an overview of good clinical practices (GCP) for clinical research and clinical trials. It discusses the definition of clinical research and clinical trials, the phases of clinical trials, important historical documents that shaped ethical standards like the Declaration of Helsinki and Nuremberg Code, and the key principles of GCP according to the WHO and ICH guidelines. These principles aim to ensure the safety and well-being of research subjects, scientific validity of the research, and compliance with regulations.











































































































