Downloaded 229 times




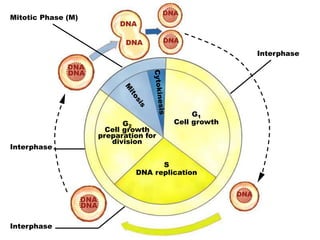
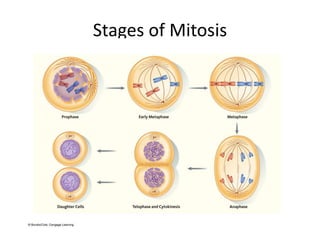


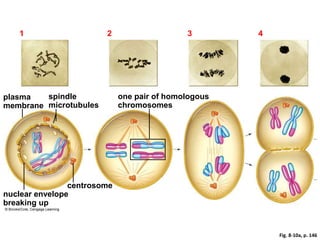

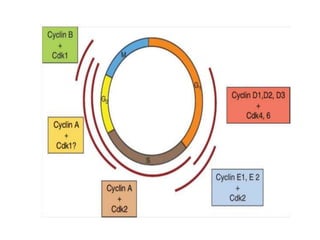

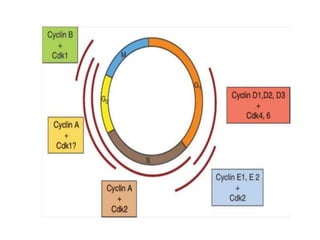
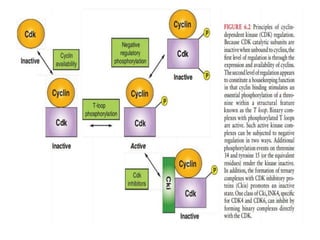




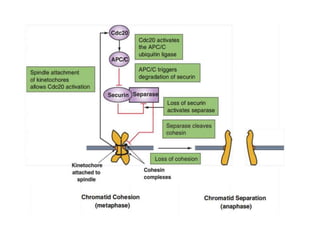


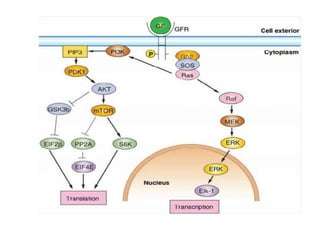

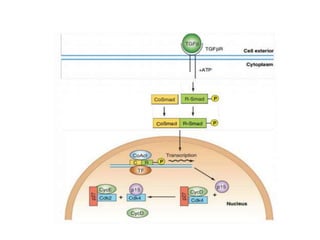


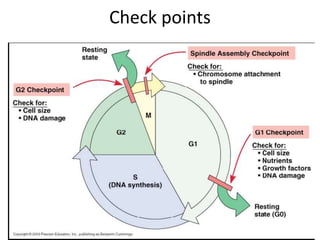








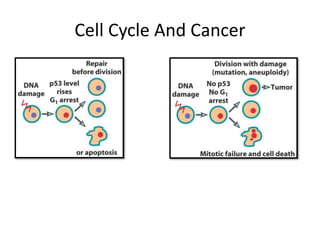






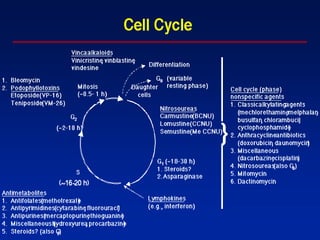
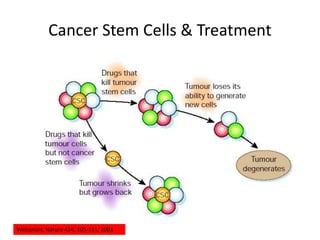


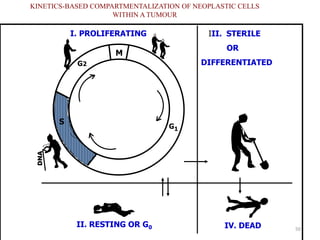


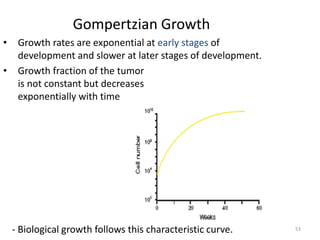
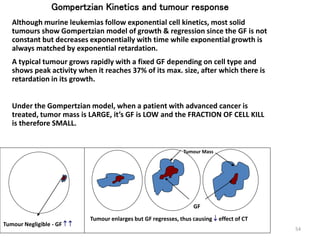








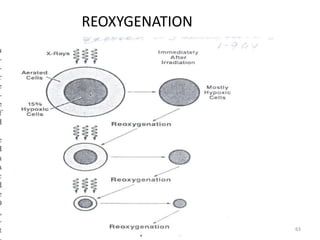



- The cell cycle consists of four main phases - G1, S, G2, and M. The G1, S, and G2 phases make up interphase. - The cell cycle is tightly regulated by cyclins and cyclin-dependent kinases (CDKs). Different cyclin-CDK complexes control progression through the different cell cycle phases. - Checkpoints exist to monitor DNA damage before progression into S phase and M phase. These checkpoints are regulated by proteins like ATM, ATR, Chk1, Chk2, and p53. - Dysregulation of cell cycle control and checkpoint pathways contributes to uncontrolled cell proliferation in cancer. Both oncogenes and tumor








![Rrecent advances in linear accelerators [MR linac]](https://cdn.slidesharecdn.com/ss_thumbnails/icroproadvance2021-recentadvancesinlinearaccelerators-211201040416-thumbnail.jpg?width=640&height=640&fit=bounds)









































![Chapter 39 role of radiotherapy in benign diseases.pptx [read only]](https://cdn.slidesharecdn.com/ss_thumbnails/chapter39roleofradiotherapyinbenigndiseases-191105205437-thumbnail.jpg?width=640&height=640&fit=bounds)




































