
Downloaded 27 times




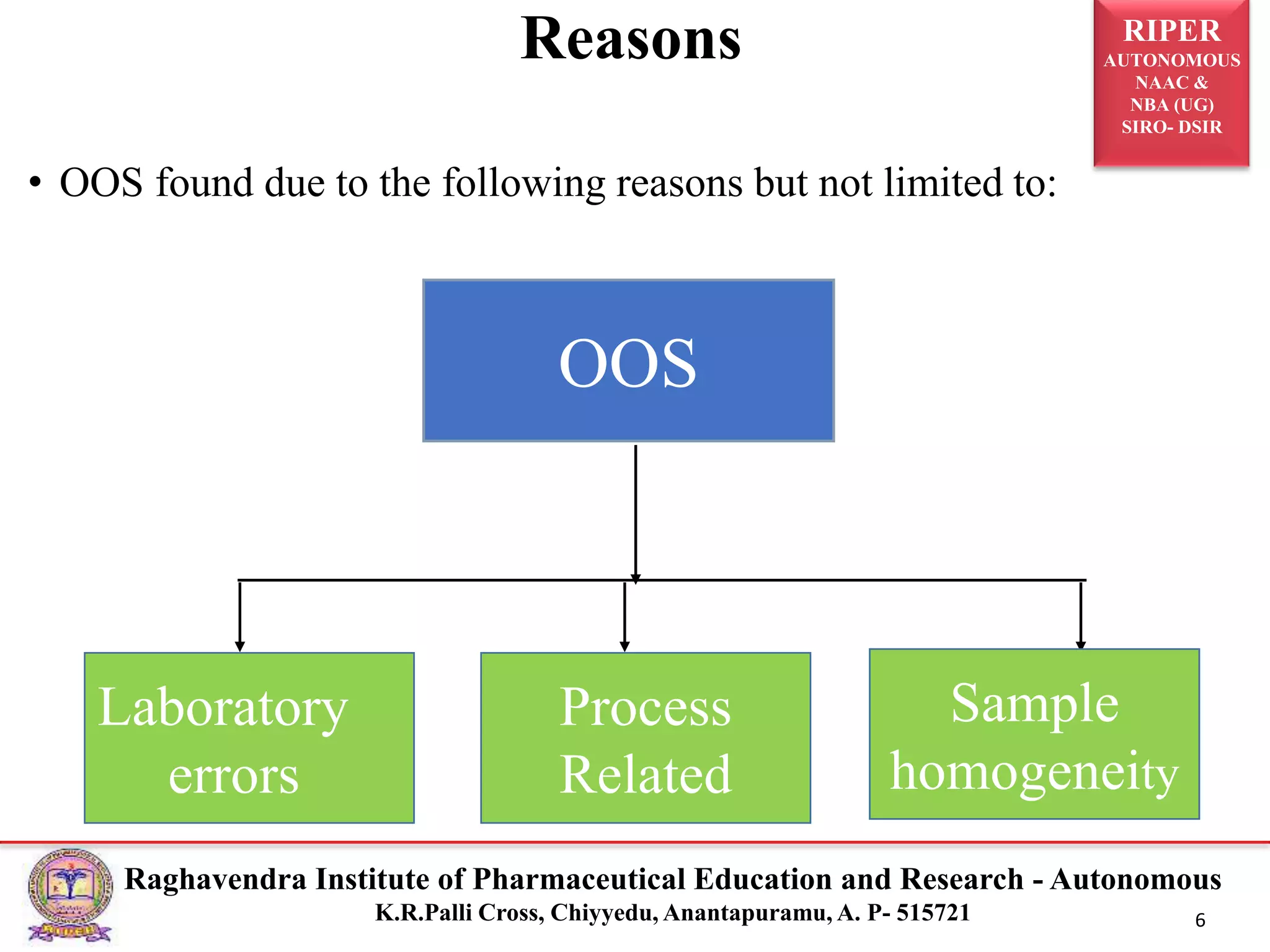
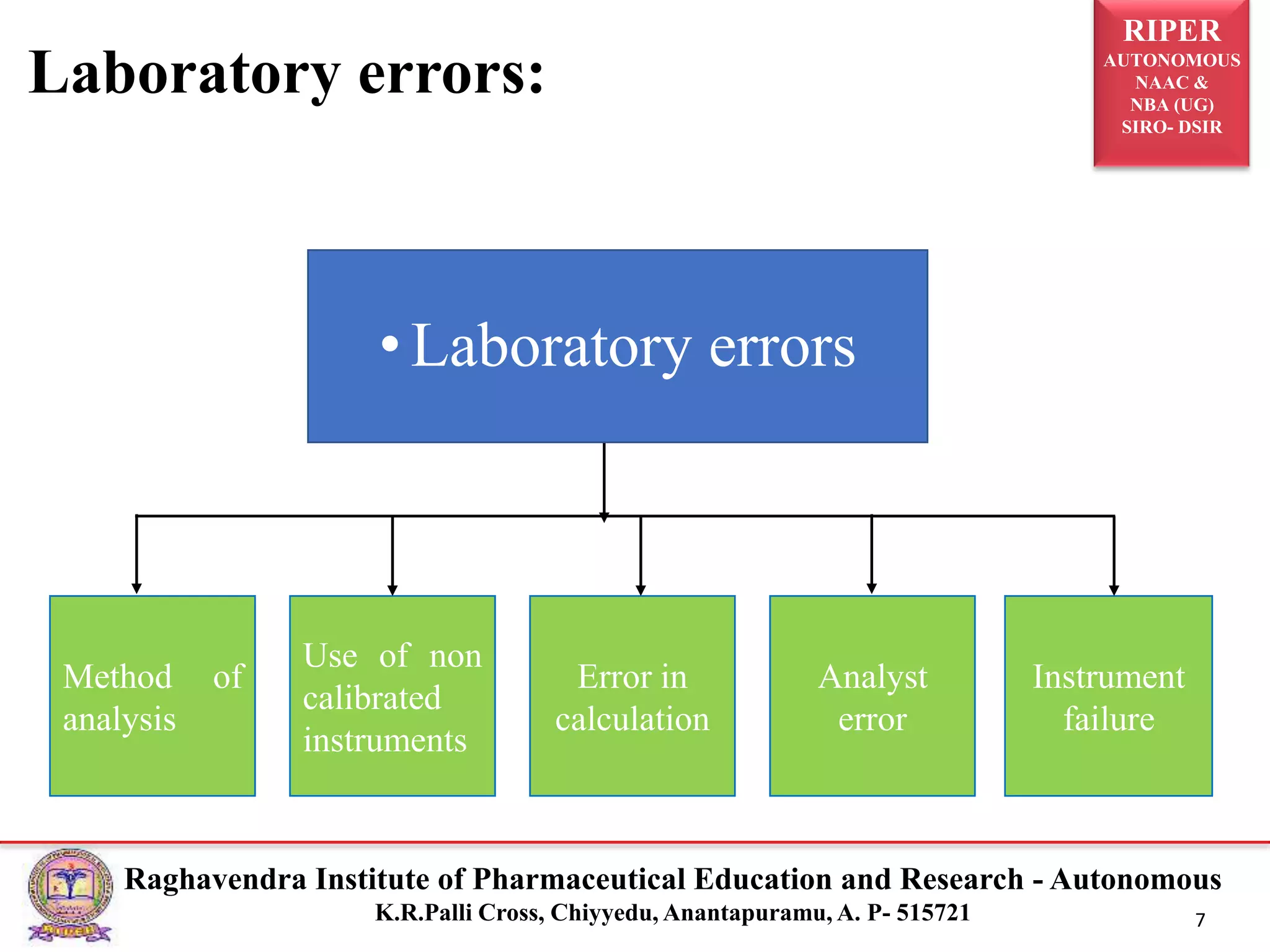
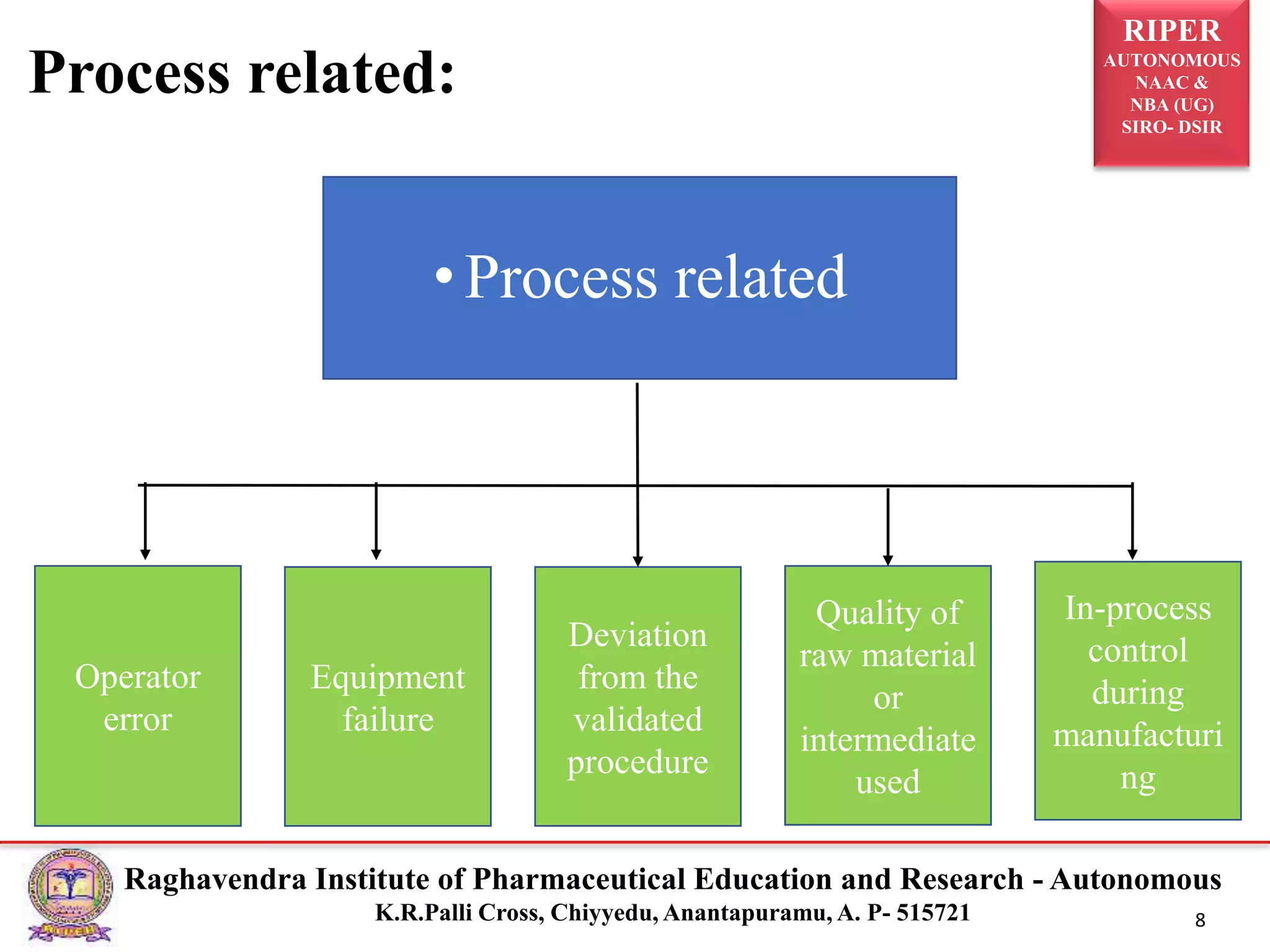
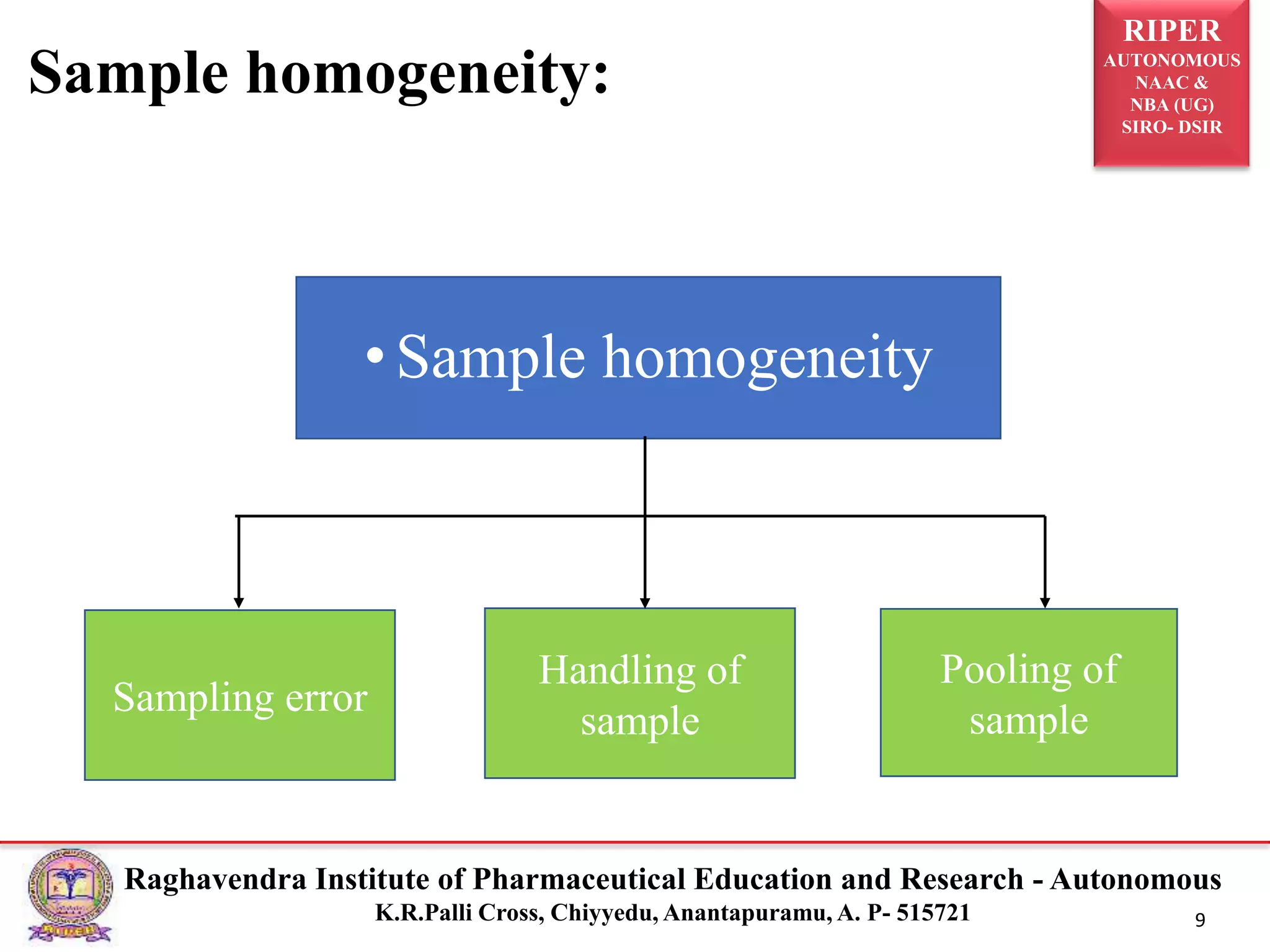

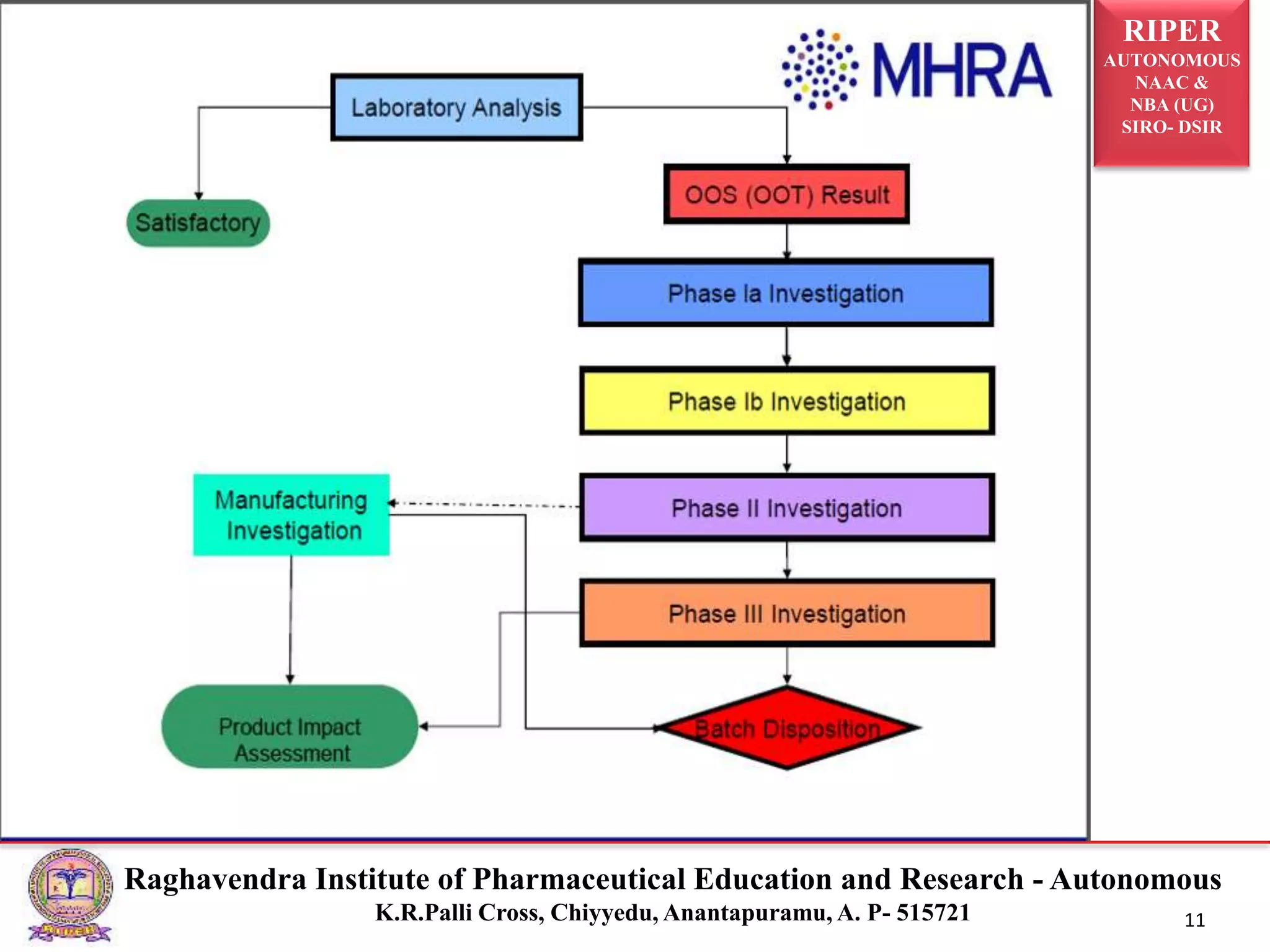










The document is a seminar report discussing Out Of Specification (OOS) results in pharmaceutical quality assurance, including definitions, investigations, and guidelines from regulatory bodies like MHRA and CDER. It highlights the reasons for OOS results, such as laboratory errors and process-related issues, and provides a case study involving Barr Laboratories that emphasizes the importance of proper testing protocols. The report concludes with key lessons on managing OOS results and the necessity of robust investigation plans.























































































