Downloaded 1,169 times
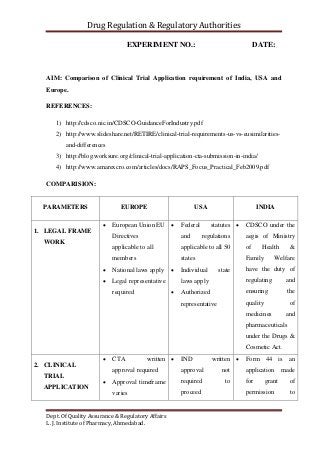

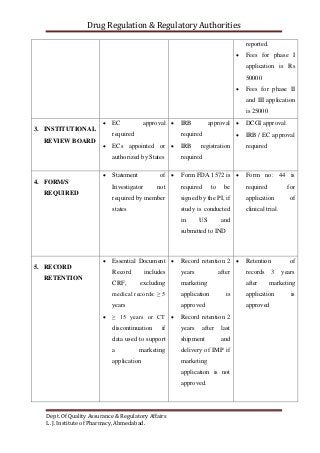
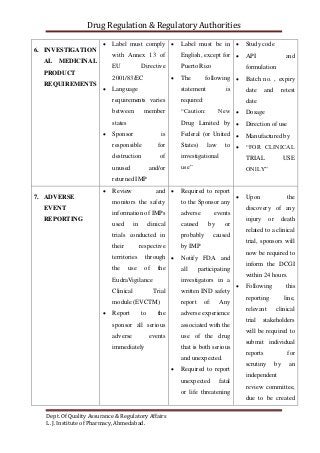
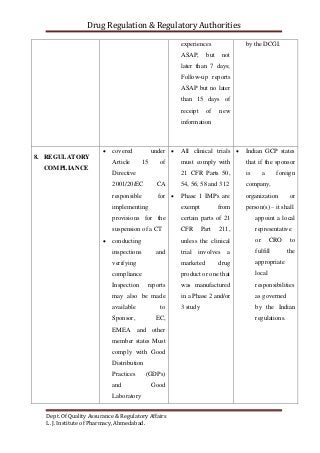


The document compares the clinical trial application requirements of India, the United States, and Europe. Some key differences include: - Europe requires approval of a clinical trial application, while the US only requires an investigational new drug application be filed. - India requires forms, documentation of chemical/toxicology data, and fees to be submitted with the application. - The US, Europe, and India all require institutional review board or ethics committee approval before starting a trial. - Reporting and retention of adverse events and trial records differs between the regions' regulations.
















![European_Union.ppt.Nikhil[1].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/europeanunion-220803170320-4be1aa31-thumbnail.jpg?width=640&height=640&fit=bounds)











































































