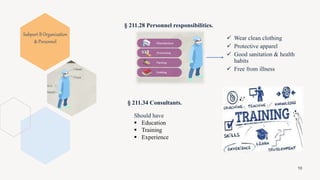
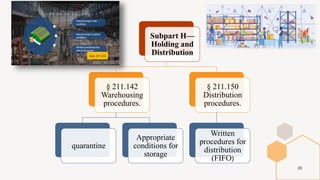
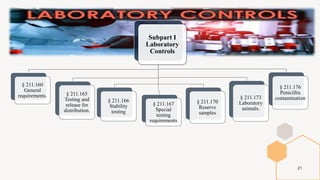
The document provides an overview and summary of 21 CFR Part 211, which establishes the current good manufacturing practice (cGMP) regulations for finished pharmaceuticals enforced by the US Food and Drug Administration (FDA). It discusses the key subparts and sections of 21 CFR Part 211, including organization and personnel, facilities, equipment, production and process controls, and laboratory controls. The presentation aims to explain the cGMP regulations and quality standards that pharmaceutical manufacturers must comply with to ensure the safety, efficacy and purity of pharmaceutical products.