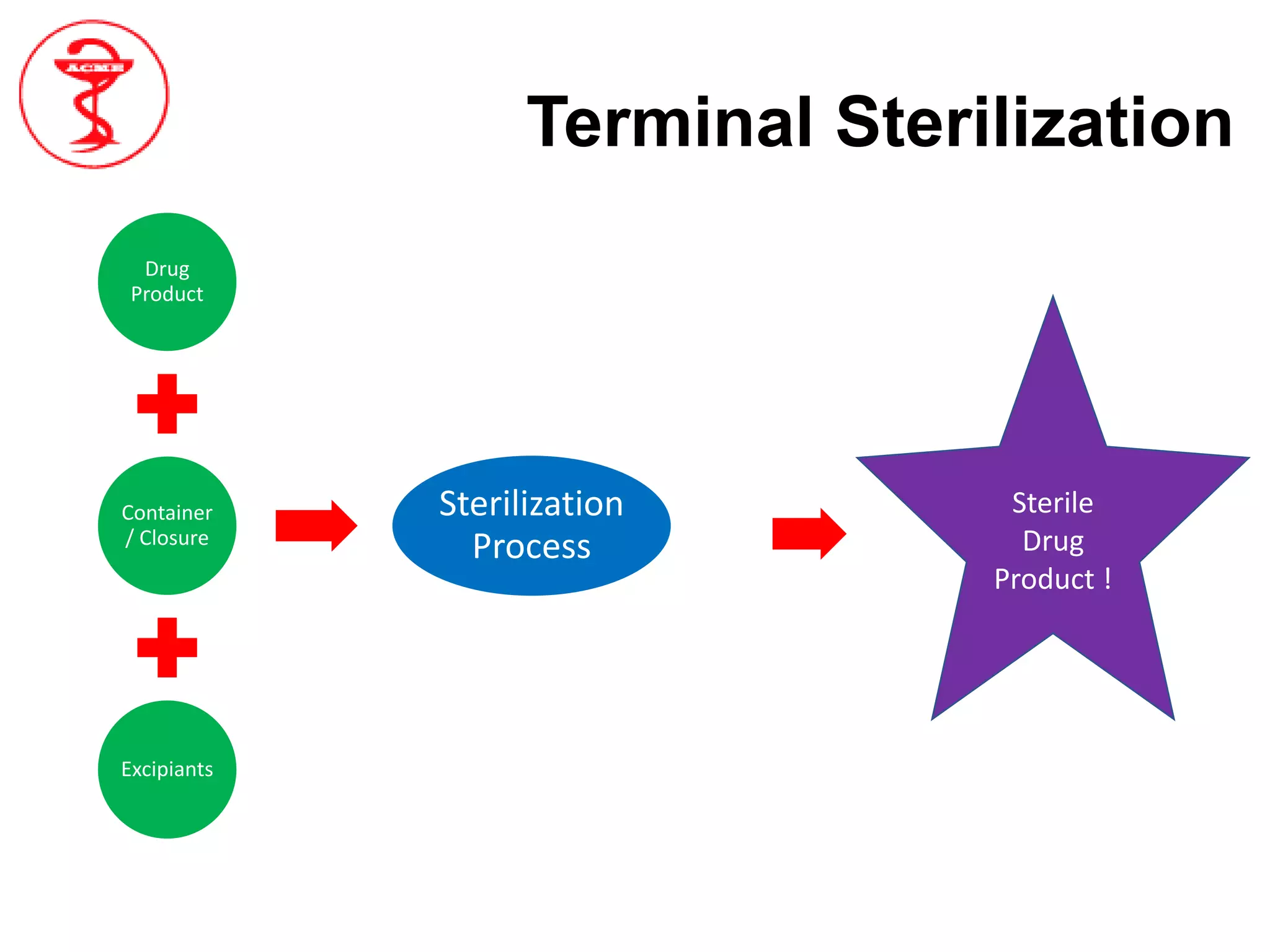
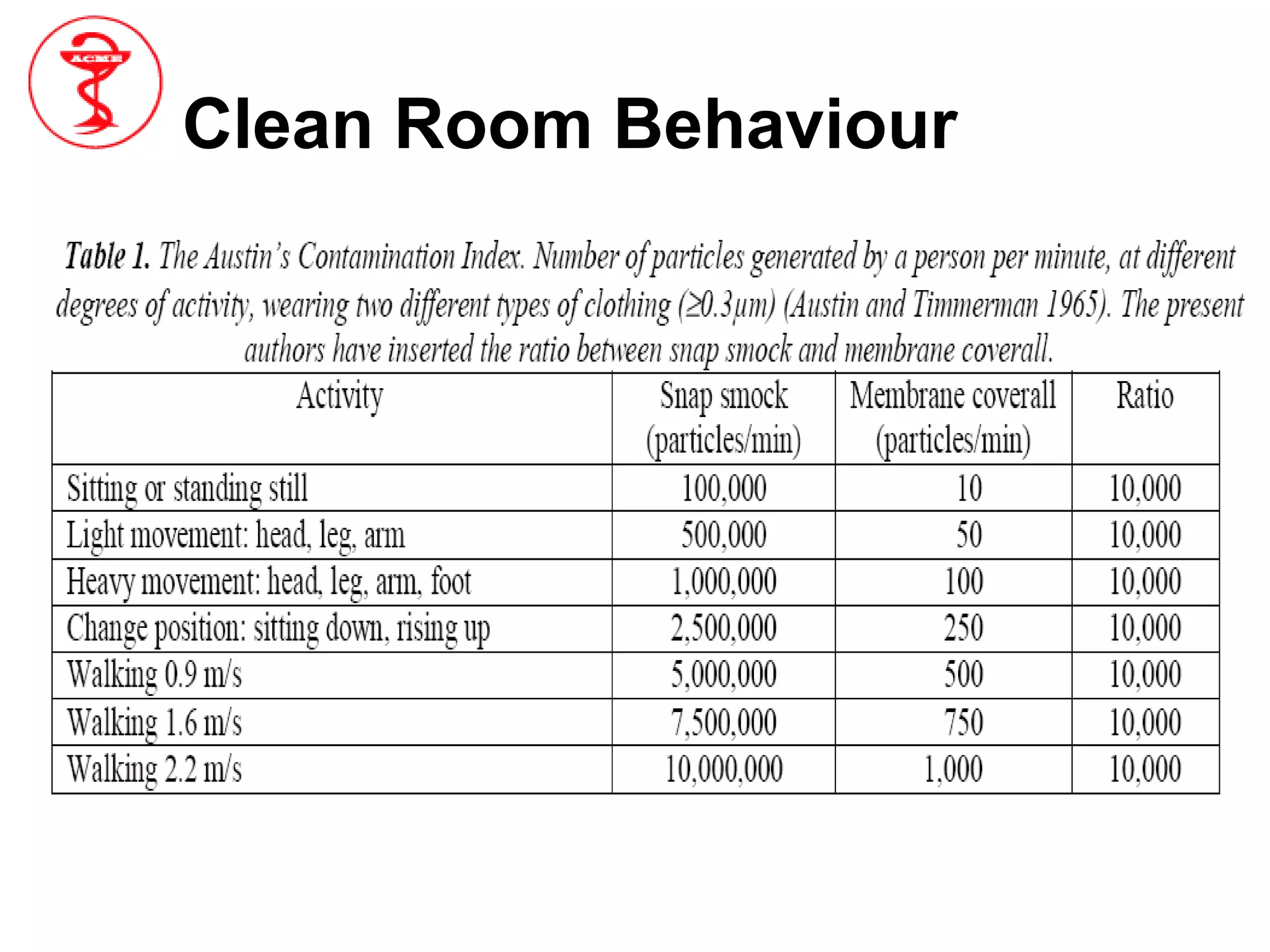
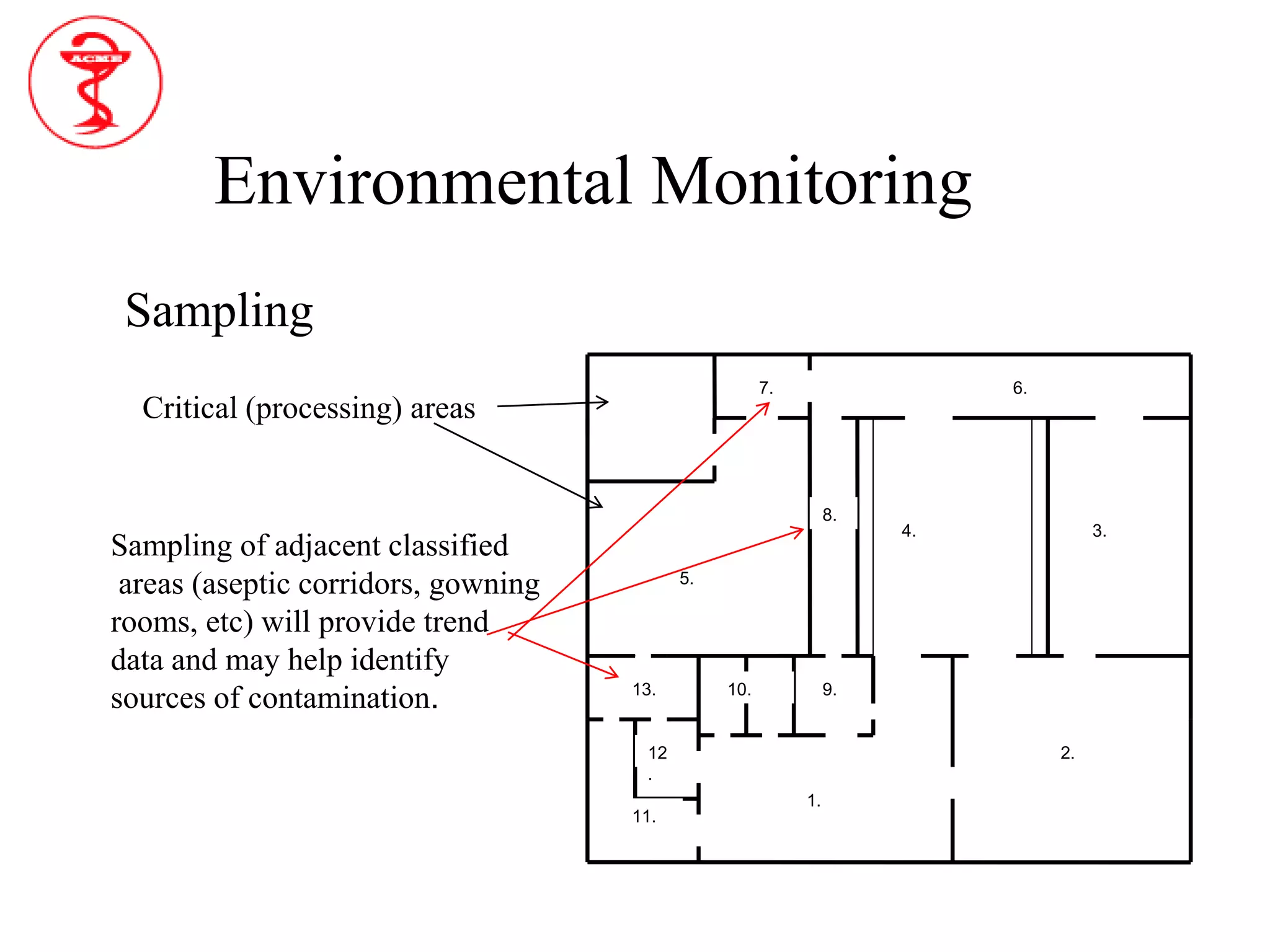
The document provides an overview of aseptic processing and contamination control. It defines aseptic processing and compares it to terminal sterilization. Sources of contamination during aseptic processing are discussed, including personnel, air, and equipment. Methods to control contamination are outlined, including quality risk management, contamination control strategies, cleaning and disinfection procedures, environmental monitoring programs, media fills, and quality control testing.