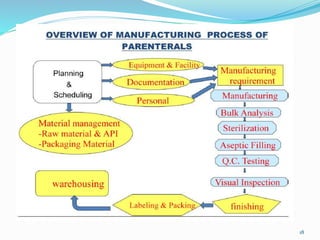
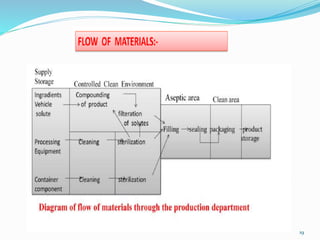
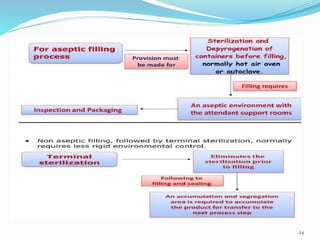
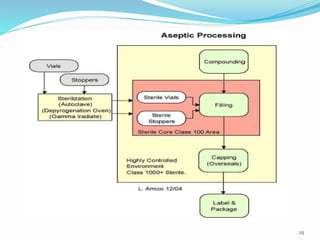
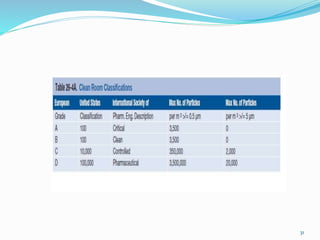
This document discusses sterile products and clean room classifications. Sterile products must be free from microorganisms and pyrogens. They include parenterals, ophthalmics, and irrigation fluids. Several factors are important for sterile compounding including clean facilities, trained personnel, and sterilization/stability principles. Clean rooms are classified based on particulate levels, with grades A through D (or classes 100 through 100,000) used in pharmaceutical facilities. Grade A/class 100 areas are needed for high-risk aseptic operations.
















![17
17
Control Action
Design airflow path to shield humans from
surroundings.
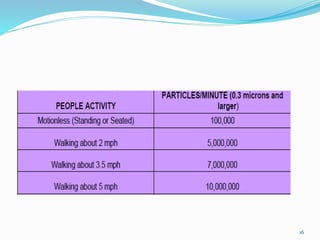
Use of air showers [to continually wash occupants
with clean air].
Using hard-surfaced, non-porous materials such as
polyvinyl panels, epoxy painted walls, & glass board
ceilings.
Proper gowning procedures, head wear mask etc.
A super clean environment with controlled
temperature & relative humidity has now become
an essential requirement for a wide range of
applications in Pharmaceutical Plants.
More critical in case of Sterile products.](https://image.slidesharecdn.com/lecture17-sterileproducts1-230708143051-4374dc5f/85/Sterile-products-pptx-17-320.jpg)