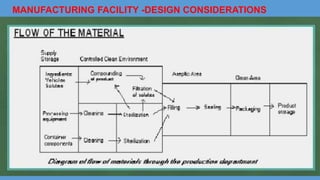
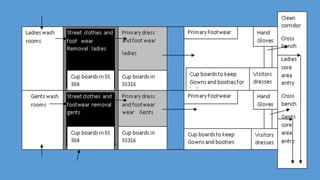
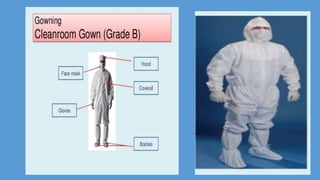
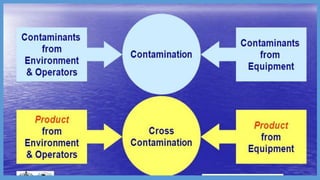
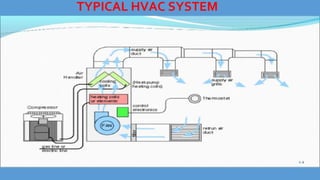
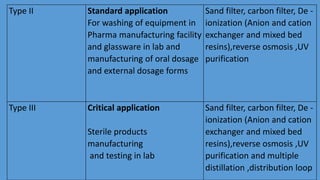
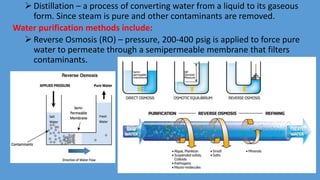
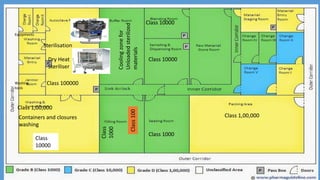
Parenteral manufacturing facilities must adhere to stringent design and operational standards to ensure product sterility and stability, focusing on factors such as clean room conditions, personnel hygiene, and environmental control. Contaminants, including microbial pathogens, pose significant risks, and effective cleaning, disinfection procedures, and proper HVAC systems are critical for minimizing cross-contamination. Compliance with good manufacturing practices (GMP) is essential, as non-adherence can lead to product adulteration and health hazards for patients.