
Downloaded 31 times






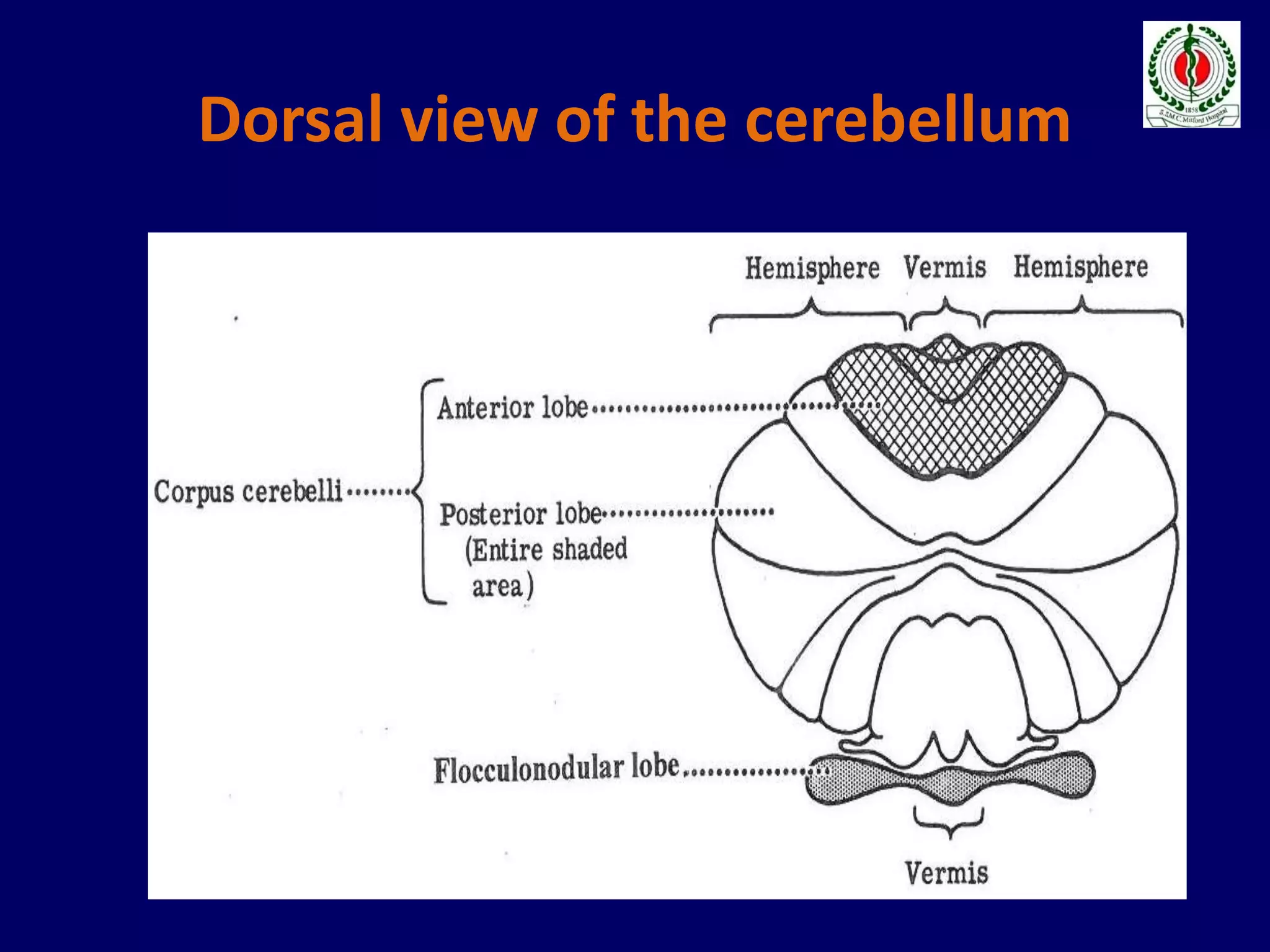

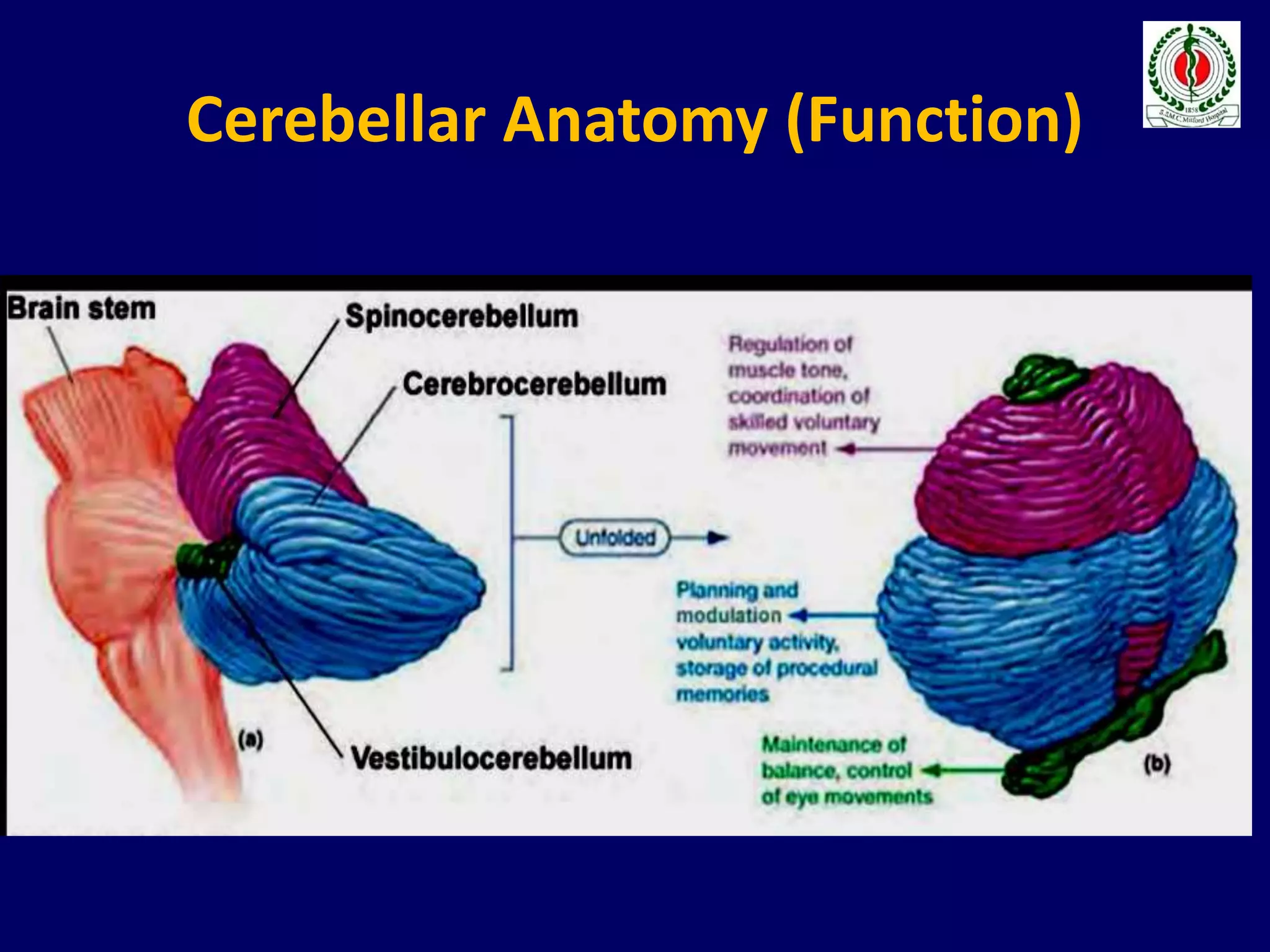





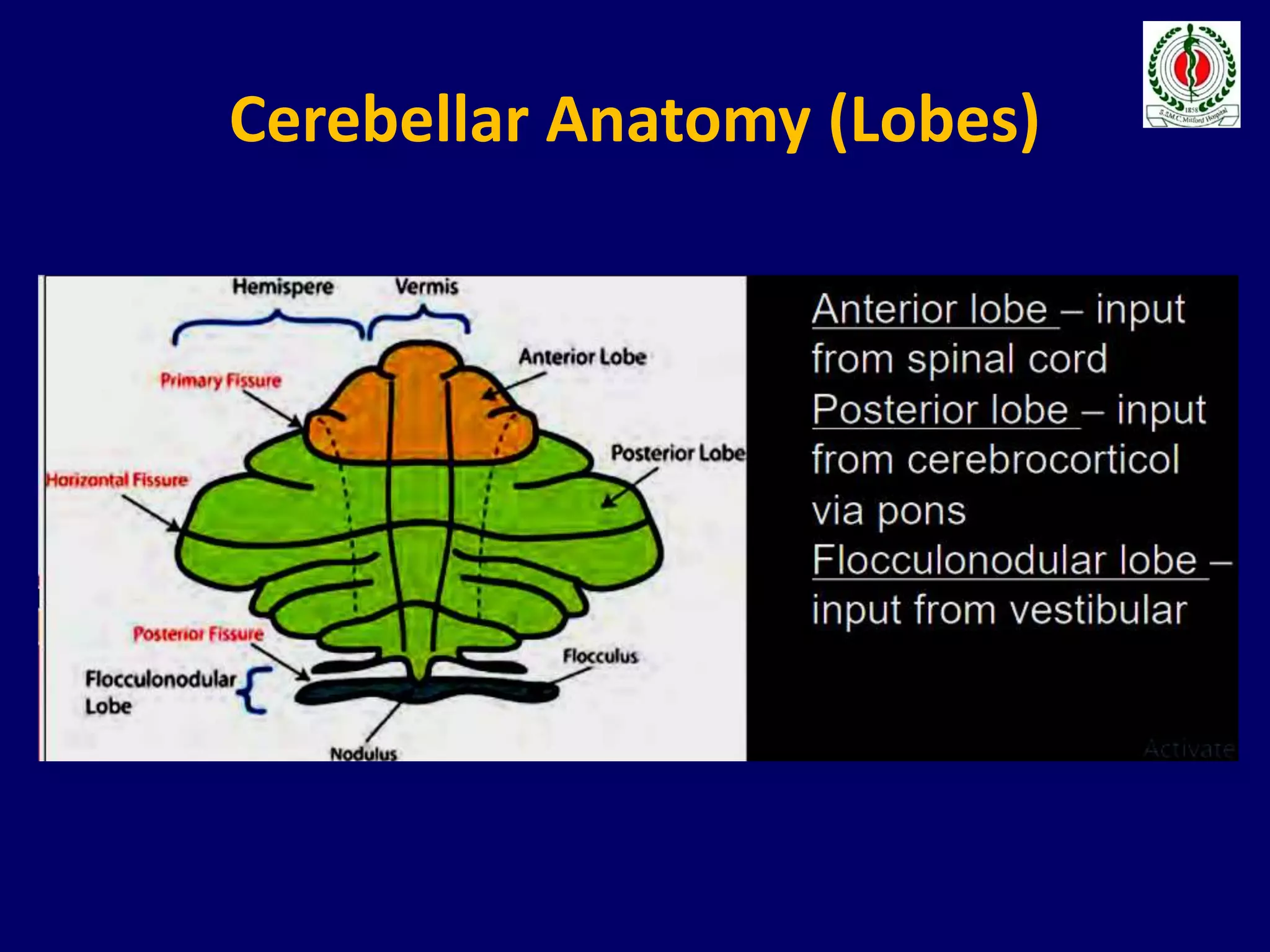
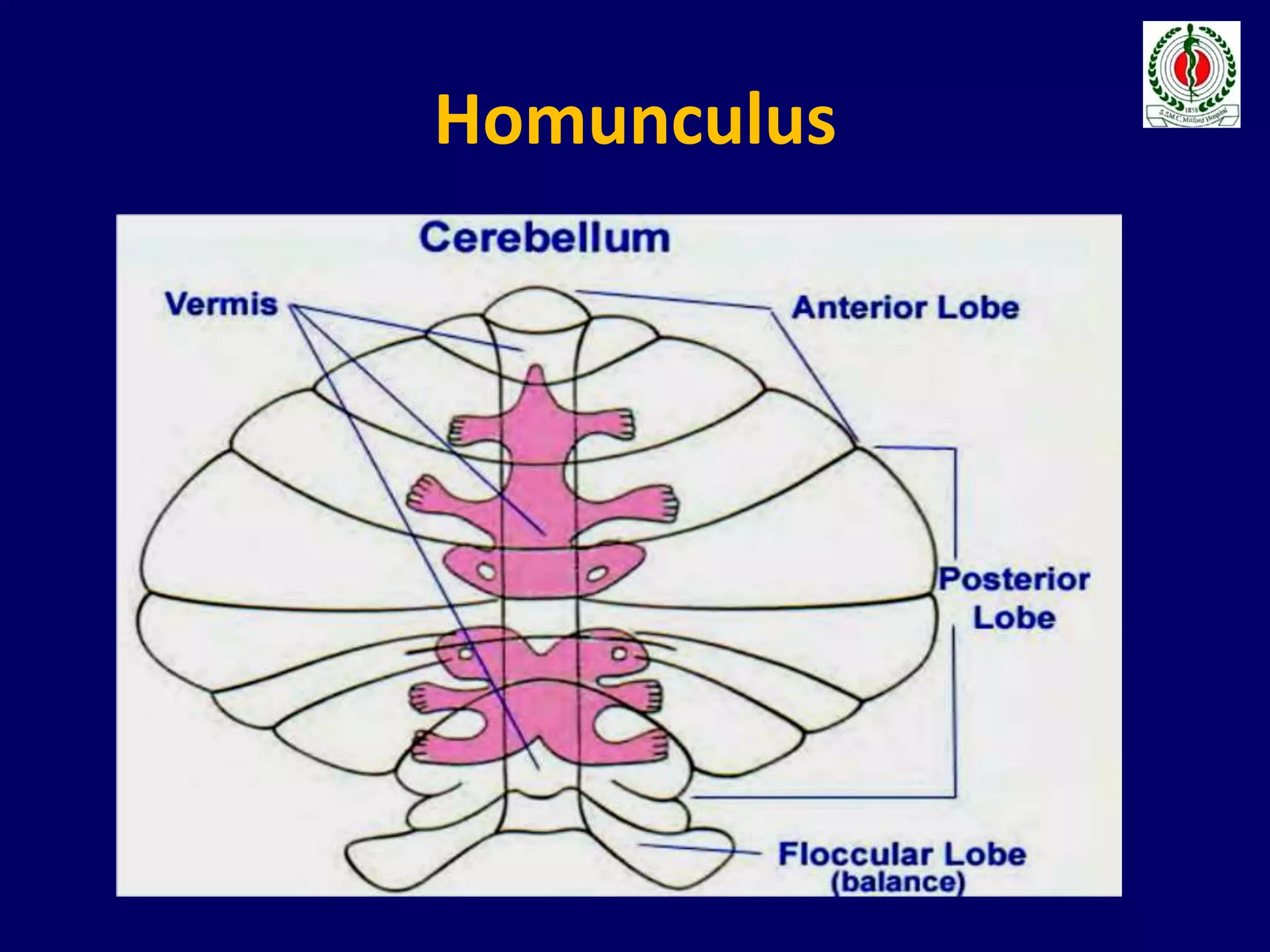
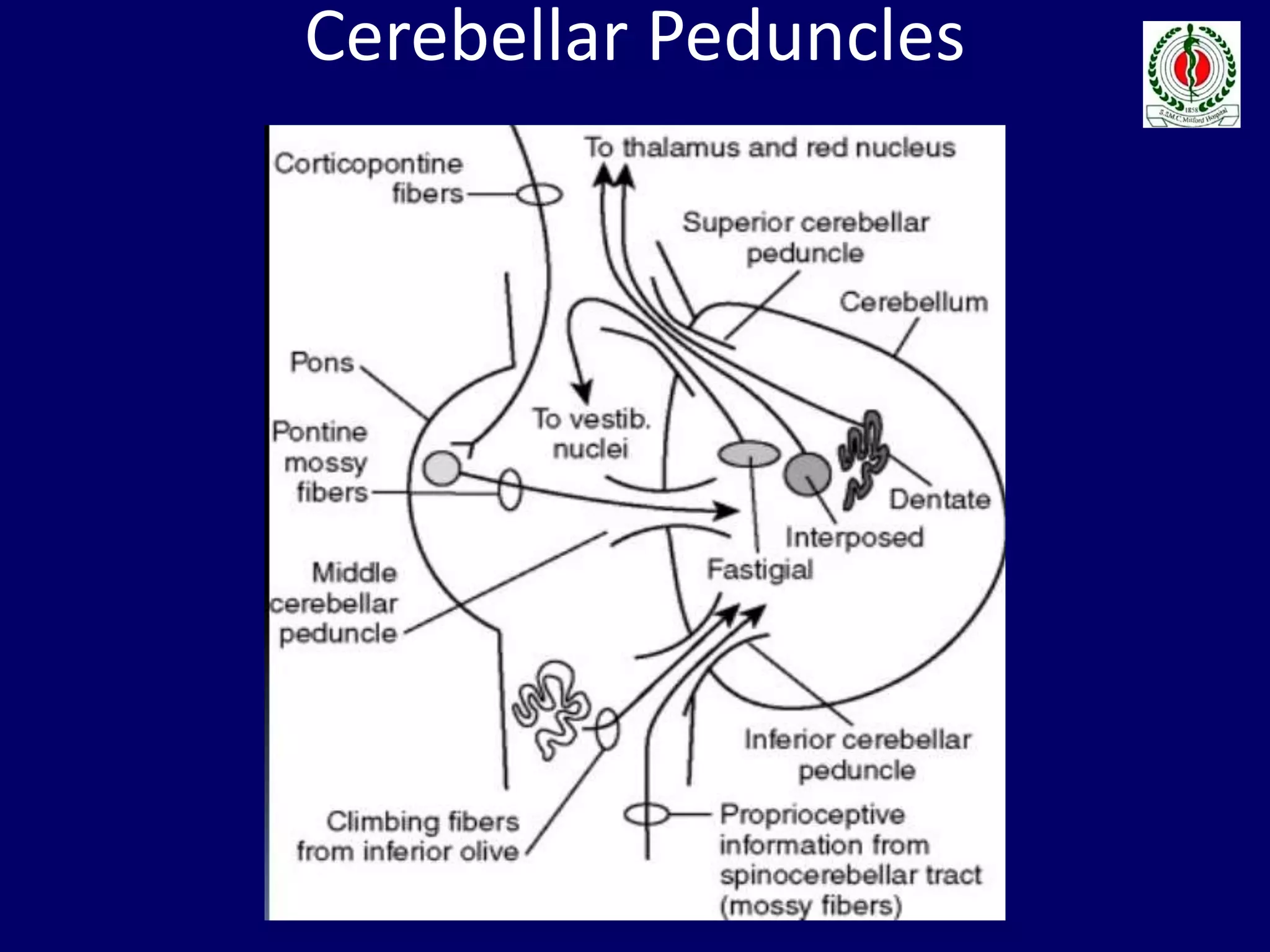
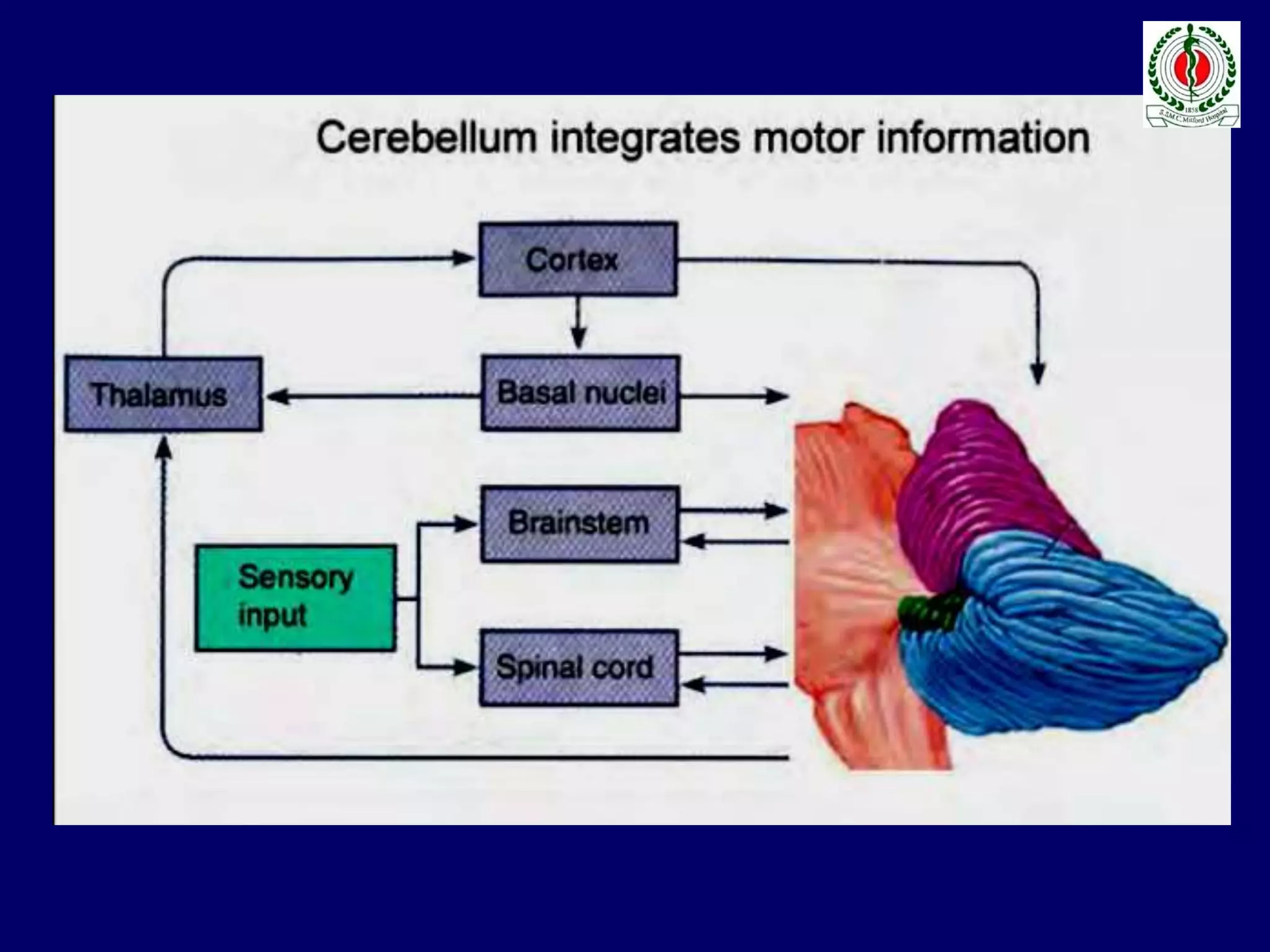
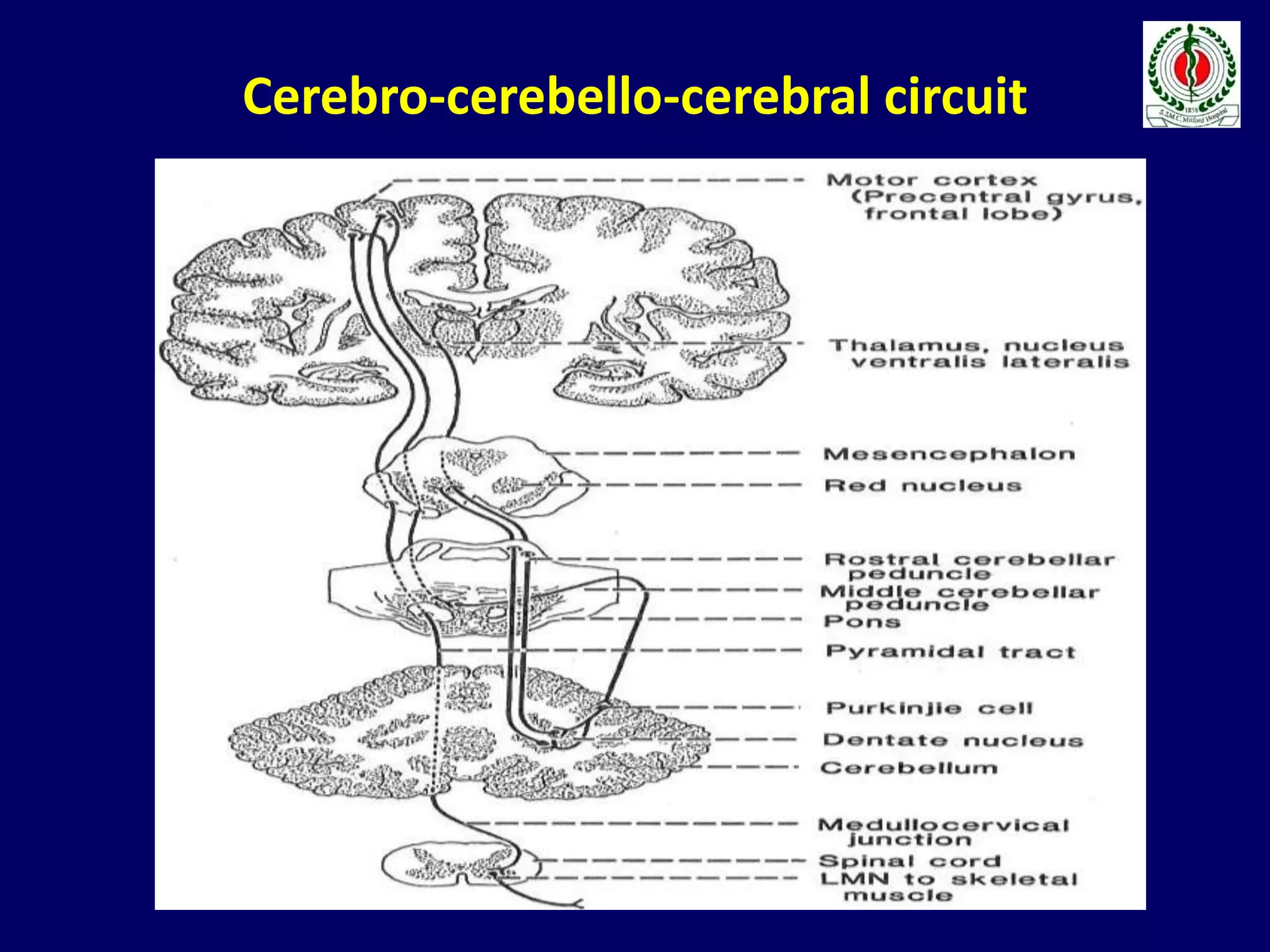


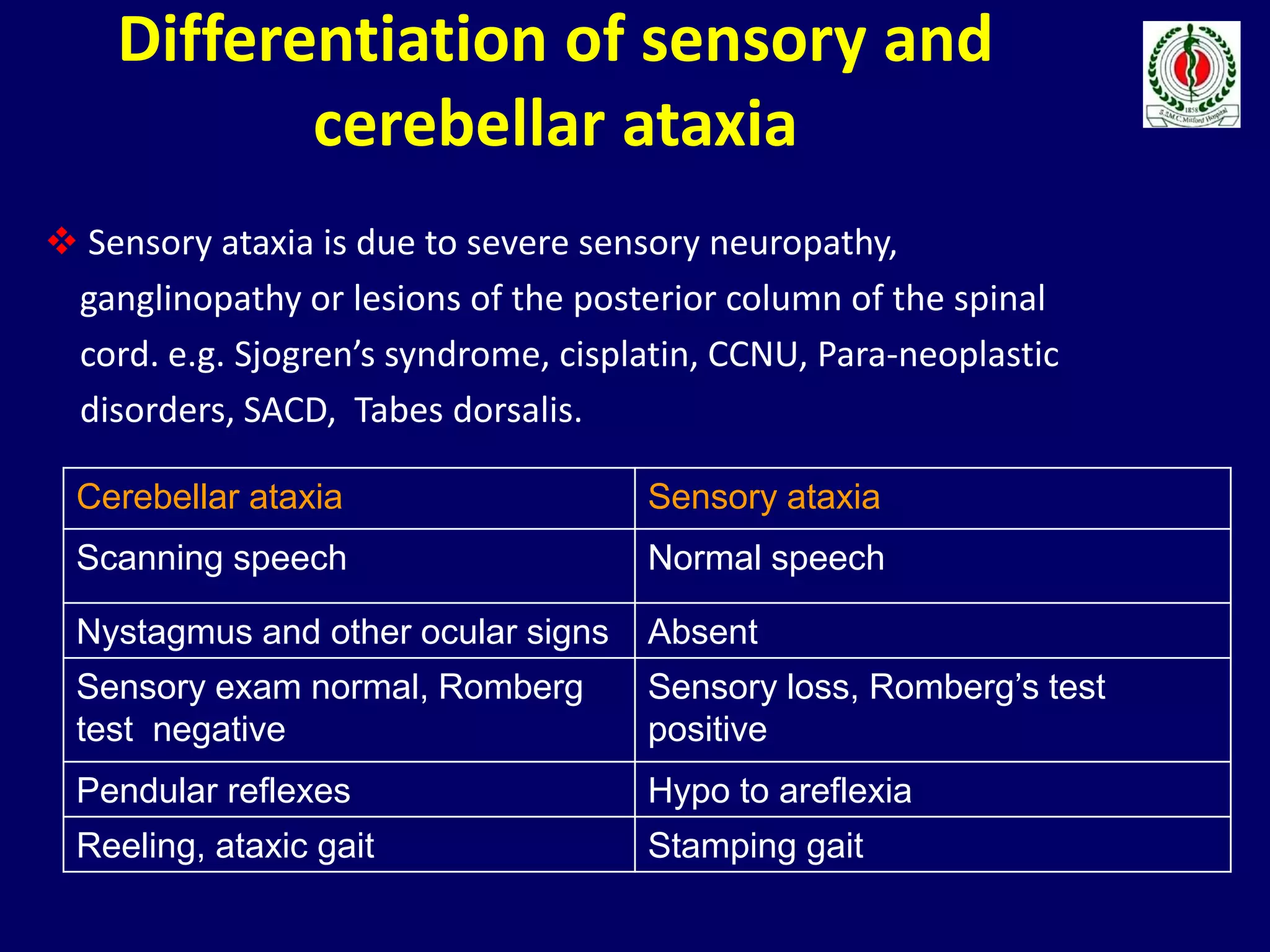


























































This document provides an overview of approach to a patient presenting with ataxia. It begins with defining ataxia and describing the anatomy and physiology of the cerebellum. It then discusses differentiating cerebellar ataxia from other causes. The document classifies ataxias as hereditary or acquired and further subclassifies each. Within hereditary ataxias, it describes several specific conditions like Friedreich's ataxia and autosomal dominant ataxias. Within acquired ataxias, it briefly discusses acute cerebellar ataxia and ataxia-telangectasia. Treatment approaches are also mentioned.









































































