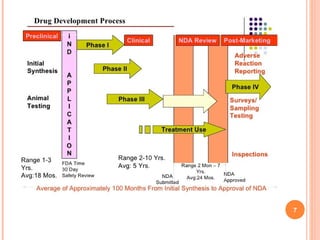
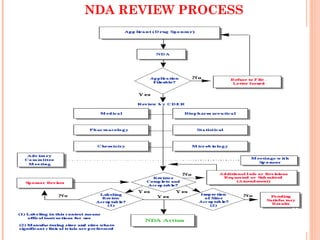
The document provides an overview of the New Drug Application (NDA) process used in the United States to gain approval for new drugs. It discusses how the NDA process has evolved since 1938 to require evidence of both safety and efficacy. It also describes the various sections required in an NDA, including summaries of clinical data, chemistry and manufacturing, labeling, and safety information. The review process for an NDA by the FDA is also summarized, including timeframes for filing and reviewing an application.