Downloaded 221 times




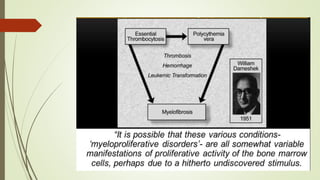



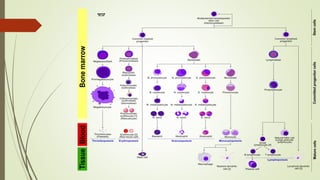


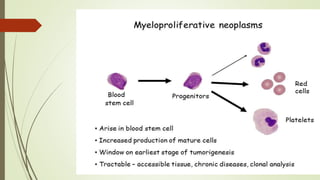
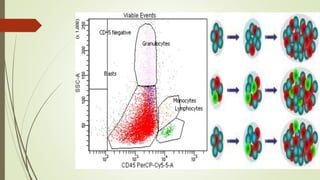

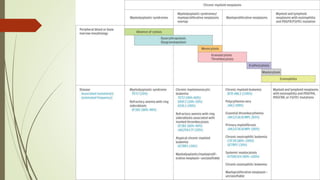


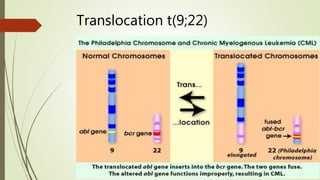



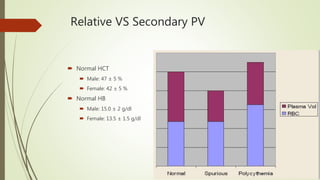
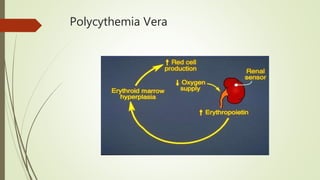



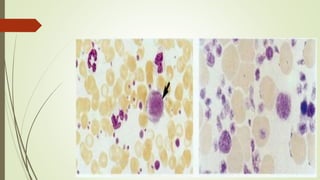


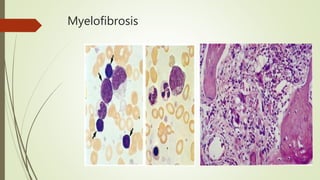


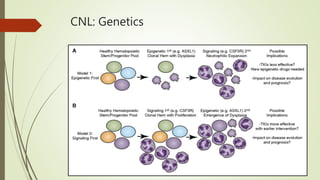


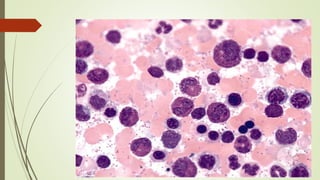


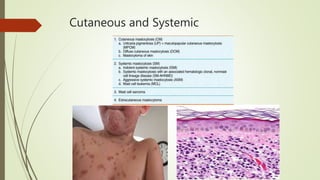



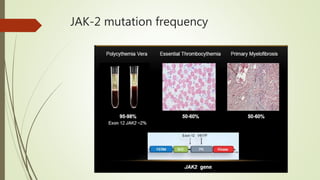
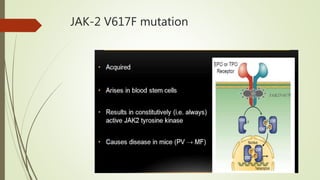
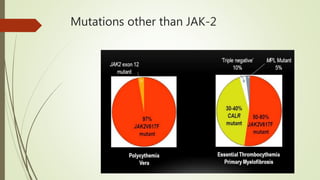

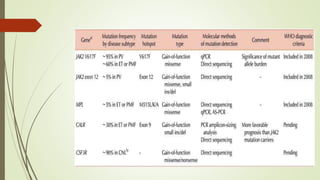


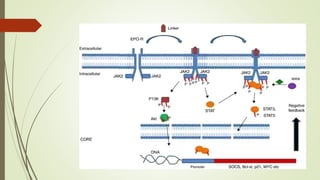
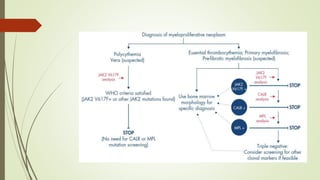
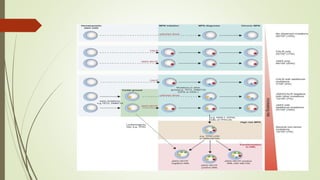

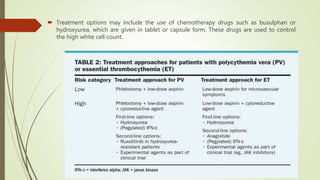





Myeloproliferative neoplasms (MPNs) are a group of disorders where the bone marrow produces too many red or white blood cells. The presentation outlines the history, classification, signs and symptoms, causes related to genetic mutations like JAK2, diagnosis through blood and bone marrow tests, and treatments including medications, radiation, surgery, and stem cell transplant. MPNs include chronic myeloid leukemia, polycythemia vera, essential thrombocythemia, myelofibrosis, and rare disorders like chronic neutrophilic leukemia and mast cell disease.



































































