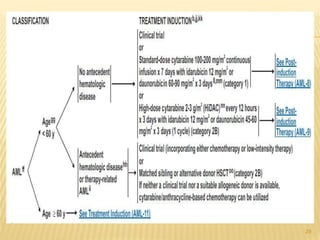
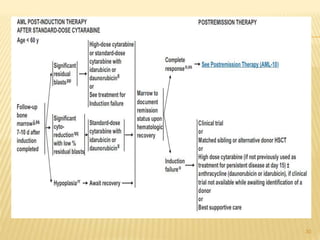
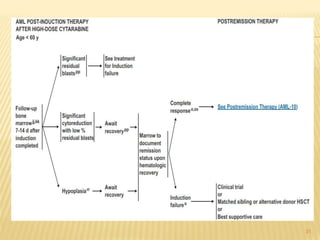
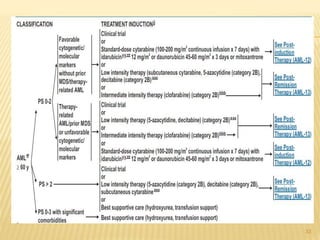
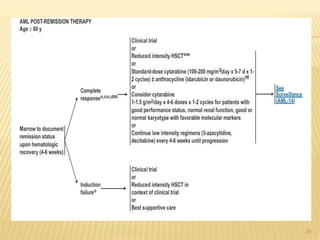
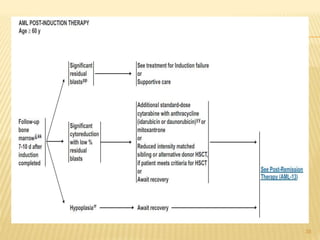
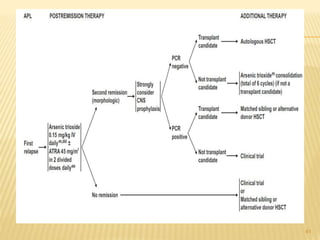
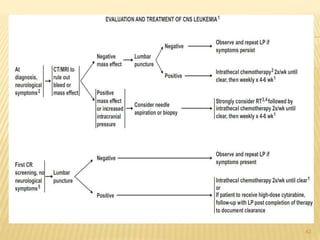
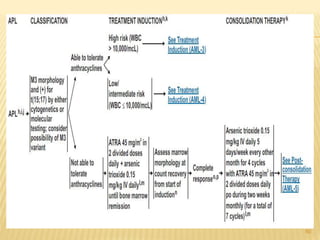
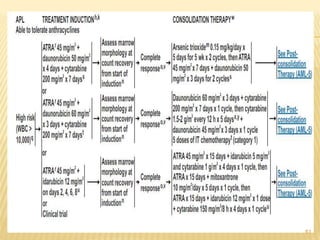
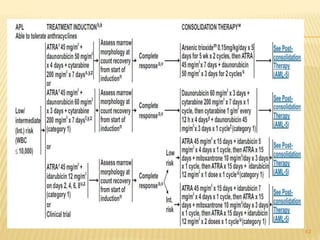
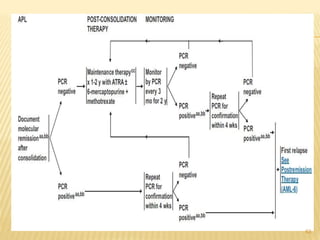
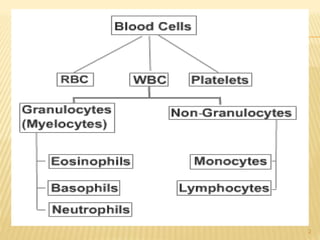
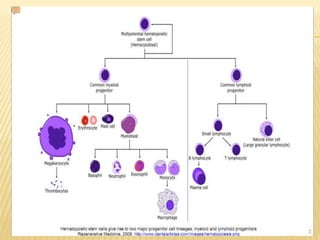
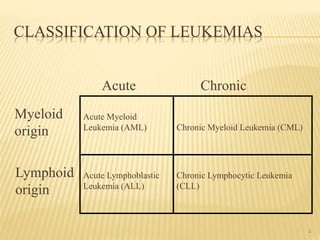
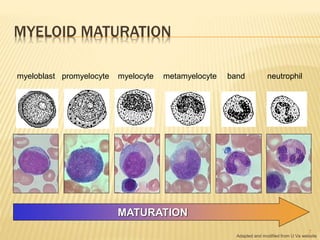
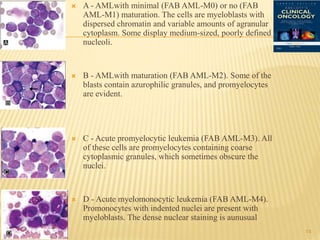
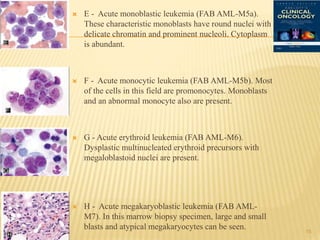
This document discusses the management of acute myeloid leukemia (AML). It begins by classifying leukemias and providing statistics on AML incidence and mortality. It then describes the clinical presentation, diagnosis, prognostic factors, treatment including induction chemotherapy and post-remission therapy, and special considerations for acute promyelocytic leukemia. The treatment sections focus on standard "7+3" induction with cytarabine and anthracyclines, achieving remission, and post-remission strategies including high-dose chemotherapy and stem cell transplant depending on risk factors.
















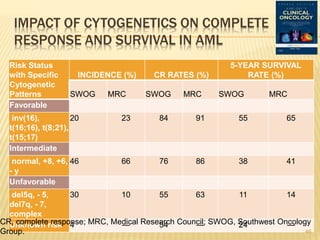
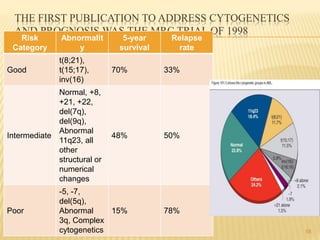
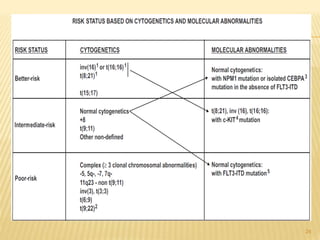
![Cytogenetic Abnormality in AML Incidence (%)[*]
Core binding factor translocations
t(8;21) 8
inv(16) or t(16;16) 9
Retinoic acid receptor translocations 10
t(15;17)
Mixed-lineage leukemia translocations
t(9;11) 2
t(10;11) 1
Other MLL translocations 3
Trisomies
+8 9
+21 3
Other trisomies 6
Deletions
- 5 (5q-) 6
- 7 (7q-) 8
- 9 (9q-) 3
Complex[†] 10
Other 17
None—normal 40 22](https://image.slidesharecdn.com/aml-management-190222092135/85/AML-management-22-320.jpg)



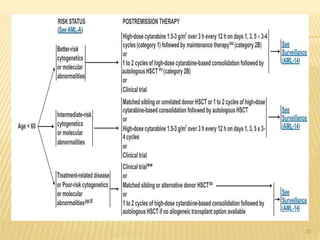
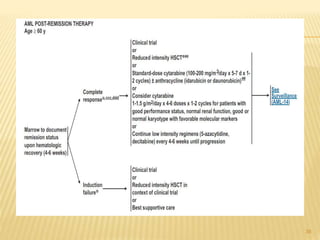
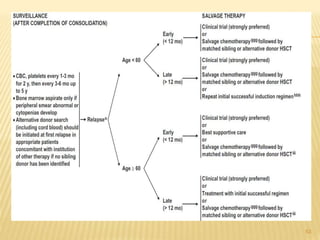
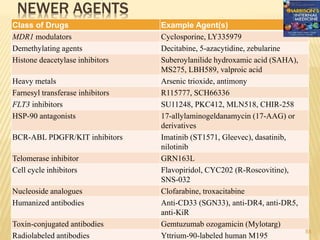
![POST REMMISION THERAPY
Postremission therapy is designed to eradicate residual leukemic
cells to prevent relapse and prolong survival.
Postremission therapy in AML is often based on age ( < 55–65
and > 55–65). For younger patients, most studies include
intensive chemotherapy and allogeneic or autologous SCT
High-dose cytarabine is more effective than standard-dose
cytarabine.
High-dose cytarabine significantly prolonged CR and increased
the fraction cured in patients with favorable [t(8;21) and inv(16)]
and normal cytogenetics, but it had no significant effect on
patients with other abnormal karyotypes
28](https://image.slidesharecdn.com/aml-management-190222092135/85/AML-management-28-320.jpg)