Міністерство охорони здоров’яУкраїни
Національна фармацевтична академія України
П.О. Безуглий, В.О. Грудько, С.Г. Леонова, С.Г. Таран,
І.В. Українець, Н.В. Гарна, В.А. Георгіянц, Л.В. Сидоренко
Посібник
з фармацевтичного аналізу
під загальною редакцією П.О. Безуглого
Харків
Видавництво НФАУ
2001
2.
3
Передмова
Фармацевтична хімія –наука, що вивчає способи одержання лікарських
речовин, їх будову, зв’язок між будовою і дією на організм, фізичні та хімічні
властивості лікарських речовин, стабільність, а також методи контролю їх
якості.
Методи контролю якості або фармацевтичний аналіз, займаючи чільне
місце при вивченні фармацевтичної хімії, має свої специфічні особливості, що
значною мірою відрізняють його від інших методів аналізу.
По-перше, це пов’язано з тим, що лікарські засоби мають різну хімічну
природу, до їх складу можуть входити декілька речовин, в багатьох випадках
хімічна будова їх дуже складна, деякі групи лікарських речовин є малостійкі
при зберіганні у невідповідних умовах.
По-друге, до фармацевтичного аналізу ставляться високі вимоги,
критеріями яких являються: точність, специфічність, чутливість. Це викликано
великою відповідальністю за результати аналізу, за якими стоїть життя
людини – хворого, який вживатиме ліки.
Названі особливості фармацевтичного аналізу потребують як ґрунтовного
знання загальних методів аналізу, так і переходу від них до багатьох
конкретних методик його проведення. Разом з тим серед них є немало
специфічних.
Враховуючи вищенаведене, автори посібника поставили задачу
узагальнити відомі методи аналізу лікарських речовин, що дасть змогу
студентам творчо підійти до виконання практичних робіт. Для досягнення
поставленої мети, а також щоб краще зв’язати теорію з практикою в кожному
розділі наводяться короткі сучасні теоретичні уявлення з фармацевтичного
аналізу в тій мірі, в якій це необхідно для свідомого його проведення і одразу
наводяться конкретні приклади аналізу окремих лікарських речовин чи
препаратів.
Значну увагу в посібнику приділено сучасним методам фізико-хімічного
аналізу – спектроскопії, хроматографії, полярографії.
Автори посібника розраховують, що він допоможе студентам ґрунтовно
засвоїти основні методи фармацевтичного аналізу, полегшить перехід від теорії
до конкретних методик практичного аналізу і будуть вдячні колегам, науковим
і практичним співробітникам за будь-які зауваження і побажання з приводу
цього видання.
3.
4
Правила роботи улабораторії фармацевтичної хімії
1. На початку кожного семестру студенти перед тим, як стати до виконання
лабораторних завдань, повинні ознайомитися з правилами роботи для
працюючих у лабораторії фармацевтичної хімії, інструкціями з техніки
безпеки та охорони праці, планом протипожежних заходів.
2. Перед початком занять чергові студенти одержують лабораторне приладдя,
яке зобов'язані здати наприкінці заняття.
3. Студенти повинні обов'язково підтримувати чистоту та порядок у
лабораторії. Працювати дозволяється тільки у халаті та спеціальному
головному уборі. На робочому столі мають знаходитися тільки предмети
необхідні для проведення досліджень.
4. Виконання лабораторного завдання дозволяється після попередньої
підготовки. Викладач контролює готовність студентів до виконання
лабораторних робіт.
5. Необхідні для проведення аналізу робочі та еталонні розчини, спеціальні
реактиви знаходяться на полицях на лабораторних столах, а титровані
розчини та хімічний посуд – у спеціальних шафах. Індикатори знаходяться
поблизу титрувальної установки. Концентровані кислоти та леткі речовини
зберігаються у витяжних шафах.
6. Перед проведенням дослідів необхідно ретельно оглянути апаратуру та
посуд, переконатися у тому, що установка або прилад зібрані правильно,
взяті хімічні речовини відповідають висунутим вимогам.
7. Відпрацьовані розчини, що містять концентровані кислоти, луги, органічні
розчинники, реактиви з отруйними речовинами та коштовними металами,
виливають у спеціальні склянки для зливу.
8. Не дозволяється виносити з лабораторії будь-які реактиви та проводити
додаткові досліди без узгодження з викладачем.
9. Після закінчення роботи ретельно вимити використаний посуд, привести у
порядок робоче місце, вимкнути газ, воду та електричні прилади.
10.Необхідно пам'ятати, що чистота, точність та кропітка праця є запорукою
успішного виконання лабораторного завдання.
Заходи безпеки під час роботи в лабораторії та способи
надання першої медичної допомоги
1. Під час роботи в лабораторії необхідно точно дотримуватись всіх заходів
безпеки згідно з правилами та інструкціями.
2. Роботу з концентрованими кислотами та іншими речовинами, які виділяють
їдкі або отруйні випари, а також речовинами і розчинами, що мають сильний
неприємний запах проводять під тягою.
3. Всі операції з діетиловим ефіром та іншими легкозаймистими речовинами
необхідно виконувати з особливою обережністю подалі від відкритого
вогню, розпечених поверхонь, електричних та електростатичних іскор.
4.
5
4. Під часроботи зі шкідливими та отруйними речовинами (солі барію,
меркурію, плюмбуму, арсену, купруму, металевою ртуттю, ціанистими
сполуками, сірководнем та ін.) треба працювати обережно, щоб ці речовини
не потрапили до організму людини. Забороняється споживання їжі в
хімічній лабораторії.
5. При порізах пересвідчитися, що в рані не залишилося уламків скла,
обробити її спиртовим розчином йоду і перев'язати.
6. При опіках зробити тривалі примочки розчином перманганату калію або
компрес зі спиртового розчину таніну.
7. При попаданні на шкіру концентрованої сірчаної кислоти необхідно
спочатку витерти уражене місце сухим ватним тампоном або салфеткою, а
потім промити великою кількістю води. При опіках шкіри, слизових
оболонок або очей кислотами спочатку добре промити уражене місце водою,
а потім – 2% розчином гідрокарбонату натрію. Для надання невідкладної
допомоги можна використати розведений у 10 разів робочий розчин натрію
карбонату.
8. При опіках їдкими лугами добре промити уражене місце водою, а потім –
1% розчином оцтової або цитринової кислоти (робочий розчин розведеної
оцтової кислоти є 30%).
9. При опіках шкіри фенолам, бромом та іншими подібними їдкими
речовинами змити уражене місце великою кількістю спирту та змастити
маззю від опіків.
10.У випадках отруєння хлором, бромом, окисами азоту та іншими подібними
речовинами слід дати потерпілому понюхати розчин аммоніаку, а потім
вивести його на свіже повітря, дати випити молока.
11.При сильних опіках, пораненнях та отруєннях, надавши першу допомогу,
потерпілого треба негайно відправити до лікарні.
12.В аптечці завжди повинен бути пepeв'язочний матеріал, розчини та
медикаменти необхідні для надання першої допомоги.
13.У разі небезпеки виникнення пожежі слід терміново перекрити подачу газу,
вимкнути витяжну вентиляцію та рубильник силової електромережі,
попередити викладача чи лаборанта і вжити заходів для ліквідації вогню.
14.Незначні осередки вогню засипають піском, накривають протипожежною
ковдрою або гасять вогонь за допомогою вогнегасника.
15.При виникненні пожежі необхідно швидко і організовано залишити
лабораторію, вивести потерпілих і надати їм першу медичну допомогу,
викликати по телефону 01 пожежну охорону.
5.
6
1. Визначення тотожностілікарських речовин
1.1. Ідентифікація неорганічних іонів
Визначення тотожності неорганічних лікарських речовин у більшості
випадків зводиться до ідентифікації окремих іонів – аніонів та катіонів, що
входять до їх складу.
В цьому розділі наведено реакції ідентифікації катіонів та аніонів, а
також деяких органічних аніонів, які входять до складу лікарських речовин.
Методики викладено відповідно до вимог діючої науково-технічної
документації. Наведено також нефармакопейні реакції, які часто
використовуються в фармацевтичному аналізі.
1.1.1. Катіони
1.1.1.1. Амоній NH4
+
1. 1 мл розчину солі амонію (0,002-0,006 г іона амонію) нагрівають з 0,5 мл
розчину натрію гідроксиду; виділяється амоніак, який виявляють за запахом
та посинінням вологого червоного лакмусового папірця:
2. 0,01-0,02 г солі амонію розчиняють в 1 краплі води, додають 1 краплю
реактиву Несслера; утворюється осад червоно-бурого кольору:
1.1.1.2. Бісмут Bi3+
1. Лікарські засоби бісмуту (близько 0,05 г іона бісмуту) збовтують з 3 мл
розведеної хлороводневої кислоти і фільтрують. До фільтрату додають 1 мл
розчину натрію сульфіду або сірководню; утворюється коричнево-чорний
осад, який розчиняється при додаванні рівного об’єму концентрованої
нітратної кислоти.
2Bi3+
+ 3S2-
→ Bi2S3↓
Bi2S3 + 8HNO3 → 2Bi(NO3)3 + 2NO↑ + 3S↓ + 4H2O
2. Лікарські засоби бісмуту (близько 0,05 г іона бісмуту) збовтують з 5 мл
розведеної сульфатної кислоти і фільтрують. До фільтрату додають 2 краплі
розчину калію йодиду; утворюється чорний осад, що розчиняється в
надлишку реактиву; з’являється жовтувато-оранжеве забарвлення:
Bi3+
+ 3I- → BiI3↓
↓ BiI3 + I- → [BiI4]-
NH4
+
+ OH
-
NH4OH NH3 + H2O
NH4
+
+ 2HgI4
2-
+ 2OH
-
NH2
Hg
Hg
I
I
+
I
-
+ 5I
-
+ 2H2O
6.
7
1.1.1.3. Ферум (II)Fe2+
1. До 2 мл розчину солі феруму (II) (близько 0,02 г іона феруму) додають
0,5 мл розведеної хлороводневої кислоти та 1 мл розчину калію фериціаніду;
утворюється синій осад, який не розчиняється у мінеральних кислотах, але
руйнується лугами з утворенням гідроксиду феруму (II):
3Fe2+
+ 2[Fe(CN)6]3-
→ Fe3[Fe(CN)6]2↓
Fe3[Fe(CN)6]2 + 6OH- → 3Fe(OH)2↓ + 2[Fe(CN)6]3-
Можливо також утворення сполуки KFe[Fe(CN)6].
2. До розчину солі феруму (II) (близько 0,02 г іона феруму) додають розчин
амонію сульфіду; утворюється чорний осад, розчинний у розведених
мінеральних кислотах:
Fe2+
+ S2-
→ FeS↓
1.1.1.4. Ферум (III) Fe3+
1. До 2 мл розчину солі феруму (III) (близько 0,02 г іона феруму) додають
0,5 мл розведеної хлороводневої кислоти та 1-2 краплі розчину калію
фероціаніду; утворюється синій осад:
4Fe3+
+ 3[Fe(CN)6]4-
→ Fe4[Fe(CN)6]3↓
Можливо також утворення осаду KFe[Fe(CN)6]. Осад розкладається лугами:
↓Fe4[Fe(CN)6]3 + 12OH- → 4Fe(OH)3↓ + 3[Fe(CN)6]4-
2. До 2 мл розчину солі феруму (III) (близько 0,001 г іона феруму) додають
0,5 мл розведеної хлороводневої кислоти та 1-2 краплі розчину амонію
роданіду; з’являється червоне забарвлення:
Fe3+
+ 3SCN- → Fe(SCN)3
3. До розчину солі феруму (III) (близько 0,001 г іона феруму) додають розчин
амонію сульфіду; утворюється чорний осад, розчинний у розведених
мінеральних кислотах:
2Fe3+
+ 3S2-
→ Fe2S3↓
1.1.1.5. Калій K+
1. До 2 мл розчину солі калію (0,01 – 0,02 г іона калію) додають 1 мл розчину
винної кислоти, 1 мл розчину натрію ацетату, 0,5 мл 95% спирту, збовтують;
поступово утворюється білий кристалічний осад, розчинний у розведених
мінеральних кислотах і розчинах лугів:
7.
8
Утворенню осаду сприяєохолодження реакційної суміші та потирання
скляною паличкою стінок пробірки.
2. До 2 мл розчину солі калію (0,005-0,01 г іона калію), попередньо прожареної
для видалення солей амонію, додають 0,5 мл розведеної оцтової кислоти та
0,5 мл розчину натрію кобальтинітриту; утворюється жовтий кристалічний
осад:
2K+
+ Na+
+ [Co(NO2)6]3-
→ K2Na[Co(NO2)6]↓
Реакцію не можна проводити в лужному або сильнокислому середовищі
внаслідок руйнування комплексного йону:
[Co(NO2)6]3-
+ 3OH- → Co(OH)3↓ + 6NO2
-
[Co(NO2)6]3-
+ 6H+
→ Co3+
+ 6HNO2
3. Сіль калію, внесена в безбарвне полум’я, забарвлює його в фіолетовий колір
або при розгляданні крізь синє скло – в пурпурово-червоний.
1.1.1.6. Кальцій Ca2+
1. До 1 мл розчину солі кальцію (0,002-0,02 г іона кальцію) додають 1 мл
розчину амонію оксалату; утворюється білий осад, нерозчинний у
розведеній оцтовій кислоті та розчині амоніаку, розчинний у розведених
мінеральних кислотах:
Ca2+
+ C2O4
2-
→ CaC2O4↓
2. Сіль кальцію, змочена хлороводневою кислотою і внесена у безбарвне
полум’я забарвлює його в цегляно-червоний колір.
COOH
CH
CH
COOH
OH
OH
CH
CH
COOK
COOH
OH
OH
+ K
+ + H
+
H
+
+ CH3COONa CH3COOH + Na
+
CH
CH
COOH
OH
OH
COOH
CH
CH
COOH
OH
OH
COOK
+ H
+
+ K
+
CH
CH
COOH
OH
OH
COOK
CH
CH
OH
OH
COOK
COO
-
+ H2O+ OH
-
8.
9
1.1.1.7. Магній Mg2+
1.До 1 мл розчину солі магнію (0,002-0,005 г іона магнію) додають 1 мл
розчину амонію хлориду, 1 мл розчину амоніаку та 0,5 мл розчину натрію
фосфату; утворюється білий кристалічний осад, розчинний у розведених
мінеральних кислотах та оцтовій кислоті:
Mg2+
+ NH4
+
+ PO4
3-
→ MgNH4PO4↓
↓MgNH4PO4 + H+
→ Mg2+
+ NH4
+
+ HPO4
2-
Амонію хлорид додається для запобігання утворення в реакційній суміші
осаду гідроксиду магнію. Надлишку амонію хлориду слід уникати, оскільки
можуть утворюватися розчинні комплекси [MgCl3]-, [MgCl4]2-.
1.1.1.8. Арсен As3+
1. До 0,3 мл розчину солі тривалентного арсену (близько 0,03 г іона арсеніту)
додають 0,5 мл розведеної хлороводневої кислоти та 2 краплі розчину
натрію сульфіду або сірководню; утворюється жовтий осад, нерозчинний в
концентрованій хлороводневій кислоті, розчинний у розчині амоніаку:
2As3+
+ 3S2-
→ As2S3↓
↓As2S3 + OH- → AsS3
3-
+ AsO3
3-
+ 3H2O
2. До 0,3 мл розчину солі тривалентного арсену (близько 0,003 г іона арсеніту)
додають 1 – 2 краплі розчину аргентуму нітрату; утворюється жовтий осад,
розчинний у розведеній нітратній кислоті та розчині амоніаку:
AsO3
3-
+ 3Ag+
→ Ag3AsO3↓
↓Ag3AsO3 + 6NH3 → 3[Ag(NH3)2]+
+ AsO3
3-
1.1.1.9. Арсен As5+
1. До 0,3 мл розчину солі п’ятивалентного арсену (близько 0,03 г іона
арсенату) додають 0,5 мл розведеної хлороводневої кислоти, 2 краплі
розчину натрію сульфіду або сірководню і нагрівають; утворюється жовтий
осад сульфіду арсену, нерозчинний у концентрованій хлороводневій кислоті,
розчинний у розчині амоніаку:
AsO4
3-
+ 2H+
+ S2-
→ AsO3
3-
+ S↓ +H2O
2AsO3
3-
+ 12H+
+ 3S2-
→ As2S3↓ + 6H2O
2. До 0,3 мл розчину солі п’ятивалентного арсену (близько 0,001 г іона
арсенату) додають 1–2 краплі розчину аргентуму нітрату; утворюється
коричневий осад, розчинний у розведеній нітратній кислоті та розчині
амоніаку:
AsO4
3-
+ 3Ag+
→ Ag3AsO4↓
↓Ag3AsO4 + 6NH3 → 3[Ag(NH3)2]+
+ AsO4
3-
3. До 0,3 мл розчину солі п’ятивалентного арсену (близько 0,001 г іона
арсенату) додають по 1 мл розчинів амонію хлориду, амоніаку та магнію
9.
10
сульфату; утворюється білийкристалічний осад розчинний у розведеній
хлороводневій кислоті (відмінність від арсенітів):
AsO4
3-
+ Mg2+
+ NH4
+
→ MgNH4AsO4↓
↓MgNH4AsO4 + H+
→ Mg2+
+ NH4
+
+ HAsO4
2-
1.1.1.10. Натрій Na+
1. 1 мл розчину солі натрію (0,01-0,03 г іона натрію), підкислюють розведеною
оцтовою кислотою, якщо необхідно, фільтрують, потім додають 0,5 мл
розчину цинк-уранілацетату; утворюється жовтий кристалічний осад:
Na+
+ Zn2+
+[(UO2)3(CH3COO)8]2-
+ CH3COO-
+ 9H2O →
→ NaZn[(UO2)3(CH3COO)9] . 9H2O↓
2. Сіль натрію, змочена хлороводневою кислотою, при внесенні в безбарвне
полум’я забарвлює його в жовтий колір.
1.1.1.11. Меркурій (II) Hg2+
1. До 2 мл розчину солі меркурію (II) (близько 0,05 г іона меркурію) додають
0,5 мл розчину натрію гідроксиду; утворюється жовтий осад:
Hg2+
+ 2OH- → HgO↓ + H2O
2. До 1 мл розчину солі меркурію (II) (0,01-0,03 г іона меркурію) обережно по
краплі додають розчин калію йодиду; утворюється червоний осад, який
розчиняється у надлишку реактиву:
Hg2+
+2I- → HgI2↓
↓HgI2 + 2I- → [HgI4]2-
1.1.1.12. Меркурій (I) Hg2
2+
1. Солі меркурію (I) утворюють з розчинами лугів чорний осад оксиду
меркурію (I):
Hg2
2+
+ 2OH- → Hg2O↓ + H2O
2. Солі меркурію (I) утворюють з розчинами йодидів жовто-зелений осад
меркурію (I) йодиду:
Hg2
2+
+ 2І- → Hg2І2↓
Осад розчиняється в надлишку реактиву з виділенням металічної ртуті:
Hg2І2↓ + 2І- → [HgI4]2-
+ Hg↓
3. З розчинами хлоридів солі меркурію (I) утворюють білий осад меркурію (І)
хлориду:
Hg2
2+
+ 2Cl- → Hg2Cl2↓
10.
11
4. При взаємодіїсолей меркурію(І) з розчином амоніаку утворюється темний
осад вільної ртуті:
Hg2Cl2 + 2NH3 →HgNH2Cl + Hg↓ + NH4Cl
1.1.1.13. Цинк Zn2+
1. До 2 мл нейтрального розчину солі цинку (0,005-0,02 г іона цинку) додають
0,5 мл розчину натрію сульфіду або сірководню; утворюється білий осад,
нерозчинний у розведеній оцтовій кислоті та легко розчинний у розведеній
хлороводневій кислоті:
Zn2+
+ S2-
→ ZnS↓
↓ZnS + 2H+
→ Zn2+
+ H2S↑
2. До 2 мл розчину солі цинку (0,005 – 0,02 г іона цинку) додають 0,5 мл
розчину калію фероціаніду; утворюється білий осад, нерозчинний у
розведеній хлороводневій кислоті:
3Zn2+
+ 2K+
+2[Fe(CN)6]4-
→ K2Zn3[Fe(CN)6]2↓
3. При взаємодії розчину солі цинку з розчином натрію гідроксиду
утворюється білий драглеподібний осад, розчинний у надлишку реактиву з
утворенням цинкатів:
Zn2+
+ 2OH- → Zn(OH)2↓
↓Zn(OH)2 + 2OH- → [Zn(OH)4]2-
1.1.1.14. Аргентум Ag+
1. До 1 мл розчину солі аргентуму (близько 0,005 г іона аргентуму) додають
розчин амоніаку до розчинення осаду, що утворюється спочатку, потім 2-3
краплі розчину формальдегіду і нагрівають; на стінках пробірки
утворюється блискучий наліт або випадає чорний осад металічного срібла:
2Ag+
+ 2OH- → Ag2O↓ + H2O
Ag2O + 4NH3 + H2O → 2[Ag(NH3)2]OH
2[Ag(NH3)2]OH + HCOH → 2Ag↓ + HCOO- + NH4
+
+ 3NH3↑ + H2O
2. 0,001 – 0,002 г іона аргентуму розчиняють в 1-2 краплях води, додають
1 краплю розведеної хлорводневої кислоти або розчину хлориду натрію;
утворюється білий сирнистий осад:
Ag+
+ Cl- → AgCl↓
Осад нерозчинний в кислотах, але легко розчиняється у розчинах амоніаку і
тіосульфату натрію:
↓AgCl + 2NH3 → [Ag(NH3)2]+
+ Сl-
↓AgCl + 2S2O3
2-
→ [Ag(S2O3)2]3-
+ Cl-
11.
12
1.1.1.15. Купрум Cu2+
1.0,002 – 0,005 г солі купруму розчиняють в 1 – 2 краплях води, додають 1
краплю розчину амоніаку; утворюється блакитний осад, який розчиняється у
надлишку розчину амоніаку; з’являється темно-синє забарвлення:
2Cu2+
+ 2OH- + SO4
2-
→ Cu2(OH)2SO4↓
↓Cu2(OH)2SO4 + 4NH4
+
+ 2OH- → 2[Cu(NH3)4]2+
+ SO4
2-
+ 4H2O
1.1.2. Аніони
1.1.2.1. Ацетати CH3COO-
1. 2 мл розчину ацетату (0,02 – 0,06 г іона ацетату) нагрівають з рівною
кількістю концентрованої сульфатної кислоти та 0,5 мл 95% спирту;
відчувається запах етилацетату:
2. До 2 мл нейтрального розчину ацетату (0,02 - 0,06 г іона ацетату) додають
0,2 мл розчину феруму (III) хлориду; з’являється червоно-буре забарвлення,
яке зникає від додавання розведених мінеральних кислот:
6CH3COO- + 3Fe3+
+ 2H2O → [Fe3(CH3COO)6(OH)2]+
+ 2H+
1.1.2.2. Бензоати C6H5COO-
1. До 2 мл нейтрального розчину бензоату (0,01 – 0,02 г іона бензоату) додають
0,2 мл розчину феруму (III) хлориду; утворюється рожево-жовтий осад,
розчинний в ефірі:
Реакцію проводять в нейтральному середовищі, бо в лужному феруму (III)
хлорид утворює бурий осад гідроксиду феруму (III), а в кислому комплексна
сіль розчиняється.
2. З розчином купруму сульфату нейтральні розчини бензоатів утворюють осад
бірюзового кольору:
CH3COOH + C2H5OH CH3C
O
OC2H5 + H2O
H2SO4
6 + 2Fe3+ + 10H2O
3
-
Fe3+ . Fe(OH)3
. 7H2O +
+ 3
COO
-
COO
COOH
2 + Cu2+
2
-
Cu2+
COO
-
COO
12.
13
1.1.2.3. Броміди Br-
1.До 1 мл розчину броміду (0,002 – 0,03 г іона броміду) додають 2 мл
розведеної хлороводневої кислоти, 0,5 мл розчину хлораміну, 1 мл
хлороформу та збовтують, хлороформний шар забарвлюється в жовто-бурий
колір:
2. До 2 мл розчину броміду (0,002 – 0,01 г іона броміду) додають 0,5 мл
розведеної нітратної кислоти та 0,5 мл розчину аргентуму нітрату;
утворюється жовтуватий сирнистий осад, нерозчинний у розведеній
нітратній кислоті та важко розчинний у розчині амоніаку:
Br- + Ag+
→ AgBr↓
↓AgBr + 2NH3 → [Ag(NH3)2]+
+ Br-
3. До 0,002 – 0,005 г броміду додають 1 краплю розчину купруму сульфату,
5 крапель концентрованої сульфатної кислоти; з’являється чорний осад; від
додавання декількох крапель води відбувається знебарвлення:
Cu2+
+ 2Br- → CuBr2↓
1.1.2.4. Йодиди I-
1. До 2 мл розчину йодиду (0,002-0,01 г іона йодиду) додають 0,5 мл
розведеної нітратної кислоти та 0,5 мл розчину аргентуму нітрату;
утворюється жовтий сирнистий осад, нерозчинний у розведеній нітратній
кислоті та розчині амоніаку:
I- + Ag+
→ AgI↓
2. До 2 мл розчину йодиду (0,003–0,02 г іона йодиду) додають 0,2 мл
розведеної сульфатної кислоти, 0,2 мл розчину натрію нітриту або розчину
феруму (III) хлориду та 2 мл хлороформу; при збовтуванні хлороформний
шар забарвлюється в фіолетовий колір:
2I- + 2NO2
- + 4H+
→ I2 + 2NO↑ + 2H2O
2I- + 2Fe3+
→ I2 +2Fe2+
3. При нагріванні 0,1 г йодиду з 1 мл концентрованої сульфатної кислоти
виділяються фіолетові пари йоду.
+ Cl
-
+ Na++ 2Br
-
+ 2H+ Br2 +
SO2N
Na
Cl
SO2NH2
13.
14
1.1.2.5. Карбонати (гідрокарбонати)CO3
2-
, HCO3
-
1. До 0,2 г карбонату (гідрокарбонату) або до 2 мл розчину карбонату
(гідрокарбонату) (1:10) додають 0,5 мл розведеної кислоти; виділяється
вуглекислий газ, який утворює білий осад при пропусканні крізь вапняну
воду:
CO3
2-
+ 2H+
→ H2CO3 → CO2↑ + H2O
HCO3
-
+ H+
→ H2CO3 → CO2↑ + H2O
Ca(OH)2 + CO2 → CaCO3↓ + H2O
2. До 0,2 мл розчину карбонату (1:10) додають 5 крапель насиченого розчину
магнію сульфату; утворюється білий осад (гідрокарбонат утворює осад
тільки при кип’ятінні):
4Mg2+
+ 5CO3
2-
+ 5H2O → 3MgCO3
. Mg(OH)2
. 3H2O↓ + 2HCO3
-
3. Розчин карбонату (1:10) при додаванні 1 краплі розчину фенолфталеїну
забарвлюється в червоний колір (відмінність від гідрокарбонатів).
1.1.2.6. Нітрати NO3
-
1. До лікарського засобу (близько 0,001 г іона нітрату) додають 2 краплі
розчину дифеніламіну; з’являється синє забарвлення:
2. До лікарського засобу (близько 0,002 – 0,005 г іона нітрату) додають по 2–3
краплі води та концентрованої сульфатної кислоти, шматочок металевої міді
і нагрівають; виділяються бурі пари двуокису азоту:
Cu + 2HNO3 + H2SO4 → 2NO2↑ + CuSO4 + 2H2O
3. Нітрати (близько 0,002 г іона нітрату) не знебарвлюють розчин калію
перманганату, підкислений розведеною сульфатною кислотою (відмінність
від нітритів).
4. До 5 – 6 крапель насиченого розчину феруму (II) сульфату додають 2 – 3
краплі розчину нітрату і перемішують, потім обережно по стінці пробірки
H
NN
N
H
N
H
N
H (конц.)
2
O O
O +
HSO4
-
H2SO4
tº
4Mg2+ + 8HCO3
-
3MgCO3
.Mg(OH)2
.3H2O + 5CO2
14.
15
приливають 5 –6 крапель концентрованої сульфатної кислоти, так аби
рідини не змішалися; на межі двох рідин з’являється буре кільце:
6Fe2+
+ 2NO3
- + 8H+
→ 6Fe3+
+ 2NO + 4H2O
FeSO4 + NO → [FeNO]SO4
1.1.2.7. Нітрити NO2
-
1. До лікарського засобу (близько 0,001 г іона нітриту) додають 2 краплі
розчину дифеніламіну; з’являється синє забарвлення (див. нітрати, 1.1.2.6.1).
2. До лікарського засобу (близько 0,03 г іона нітриту) додають 1 мл розведеної
сульфатної кислоти; віділяються жовто-бурі пари (відмінність від нітратів).
2NO2
- + 2H+
→ HNO2 → NO↑ + NO2↑ + H2O
2NO + O2 → 2NO2↑
3. Декілька кристалів антипірину розчиняють у фарфоровій чашці в 2 краплях
розведеної хлороводневої кислоти, додають 2 краплі розчину нітриту
(близько 0,001 г іона нітриту); з’являється зелене забарвлення (відмінність
від нітратів):
4. До 4-5 крапель розчину нітриту додають таку саму кількість розведеної
сульфатної кислоти і по краплі приливають розчин калію перманганату;
відбувається знебарвлення розчину калію перманганату:
5NO2
- + 2MnO4
- + 6H+
→ 5NO3
- + 2Mn2+
+ 3H2O
5. Так само як і нітрати, нітрити дають реакцію з феруму (II) сульфатом (див.
нітрати, 1.1.2.6.4). На відміну від нітратів реакція може протікати в
слабокислому середовищі.
1.1.2.8. Саліцилати C6H4(OH)COO-
1. До 2 мл нейтрального розчину саліцилату (0,002-0,01 г іона саліцилату)
додають 2 краплі розчину феруму (III) хлориду; з’являється синьо-фіолетове
або червоно-фіолетове забарвлення, яке зберігається при додаванні
невеликої кількості розведеної оцтової кислоти, але зникає при додаванні
розведеної хлороводневої кислоти. При цьому утворюється білий
кристалічний осад саліцилової кислоти:
+ H2O+ NO2
-
+ H+
N
N
CH3
CH3
O
C6H5
ON
N
N
CH3
CH3
H
O
C6H5
C
ONa
O
OH FeO
O
O
C
+ NaCl + HClCl
-
+
FeCl3+
15.
16
2. З розчиномкупруму сульфату нейтральні розчини саліцилатів утворюють
розчин зеленого кольору:
1.1.2.9. Сульфати SO4
2-
1. До 2 мл розчину сульфату (0,005 – 0,05 г іона сульфату) додають 0,5 мл
розчину барію хлориду; утворюється білий осад, нерозчинний у розведених
мінеральних кислотах:
SO4
2-
+ Ba2+
→ BaSO4↓
Реакція проводиться в солянокислому середовищі для розчинення осадів
фосфатів та карбонатів барію, які на відміну від барію сульфату розчинні у
розведеній хлороводневій кислоті.
1.1.2.10. Сульфіти SO3
2-
1. До 2 мл розчину сульфіту (0,01 – 0,03 г іона сульфіту) додають 2 мл
розведеної хлороводневої кислоти та збовтують; поступово виділяється
сірчистий газ, який виявляють за характерним різким запахом:
SO3
2-
+ 2H+
→ SO2↑ + H2O
2. До 2 мл розчину сульфіту (0,002 – 0,02 г іона сульфіту) додають 0,5 мл
розчину барію хлориду; утворюється білий осад, розчинний у розведеній
хлороводневій кислоті (відмінність від сульфатів):
SO3
2-
+ Ba2+
→ BaSO3↓
3. При додаванні до розчину сульфіту декількох крапель 0,1 моль/л розчину
йоду відбувається його знебарвлення:
SO3
2- + I2 + H2O → 2I- + SO4
2-
+ 2H+
+ Cu2+ + H+
OH
Cu
O
COOCOO
-
C
O
OH
OH
+
Cl
-
FeO
O
O
C
+ FeCl3+ 2HCl
16.
17
1.1.2.11. Тартрати (HOCHCOO-)2
1.До 1 мл розчину тартрату (близько 0,02 г іона тартрату) додають кристалик
калію хлориду, 0,5 мл 95% спирту; утворюється білий кристалічний осад,
розчинний у розведених мінеральних кислотах та розчинах лугів.Реакцію
проводять у присутності натрію ацетату при охолодженні та потиранні
скляною паличкою стінок пробірки (див. калій, 1.1.1.5.1).
2. 0,25 мл розчину тартрату (близько 0,005 г іона тартрату) нагрівають з 1 мл
концентрованої сульфатної кислоти та декількома кристалами резорцину;
через 15-30 с з’являється вишнево-червоне забарвлення.
1.1.2.12. Фосфати PO4
3-
1. До 1 мл розчину фосфату (0,01-0,03 г іона фосфату), нейтралізованого до pH
близько 7,0, додають декілька крапель розчину аргентуму нітрату;
утворюється жовтий осад, розчинний у розведеній нітратній кислоті та
розчині амоніаку:
PO4
3-
+ 3Ag+
→ Ag3PO4↓
↓Ag3PO4 + 3H+
→ 3Ag+
+ H3PO4
↓Ag3PO4 + 6NH3 → 3[Ag(NH3)2]+
+ PO4
3-
2. До 1 мл розчину фосфату (0,01-0,03 г іона фосфату) додають 1 мл розчину
амонію хлориду, 1 мл розчину амоніаку та 0,5 мл розчину магнію сульфату;
утворюється білий кристалічний осад, розчинний у розведених мінеральних
кислотах (див. магній, 1.1.1.7.1).
3. До 1 мл розчину фосфату (0,01-0,03 г іона фосфату) в розведеній нітратній
кислоті додають 2 мл розчину амонію молібдату і нагрівають; утворюється
жовтий кристалічний осад, розчинний у розчині амоніаку:
PO4
3-
+ 3NH4
+
+ 12MoO4
2-
+ 24H+
→ (NH4)3PO4
. 12MoO3↓ + 12H2O
1.1.2.13. Хлориди Cl-
1. До 2 мл розчину хлориду (0,002-0,01 г іону хлориду) додають 0,5 мл
розведеної нітратної кислоти і 0,5 мл розчину аргентуму нітрату;
утворюється білий сирнистий осад нерозчинний у розведеній нітратній
кислоті і розчинний у розчині амоніаку:
Для солей органічних основ дослідження розчинності осаду аргентуму
хлориду проводять після відфільтровування і промивання осаду водою
(якщо цього не зробити може утворитися осад органічної основи).
[Ag(NH3)2]Cl + H2O+ 2NH4OHAgCl
Cl
-
+ Ag+ AgCl
17.
18
1.1.2.14. Цитрати
1. До1 мл нейтрального розчину цитрату (0,002-0,01 г іона цитрату) додають
1 мл розчину кальцію хлориду; розчин залишається прозорим; при
кип’ятінні утворюється білий осад, розчинний у розведеній хлороводневій
кислоті:
2. До лікарського засобу (0,001-0,002) г іона цитрату додають 0,5 мл оцтового
ангідриду і нагрівають; через 20-40 с з’являється червоне забарвлення.
3. До 3-5 крапель розчину цитрату (0,0001 г іона цитрату) додають 3-5 крапель
0,01 моль/л розчину калію перманганату, 3-5 крапель насиченої бромної
води і злегка підігрівають. Після охолодження надлишок брому і оксид
марганцю (IV), що іноді утворюється, руйнують додаванням твердої
сульфосаліцилової кислоти. Випадає білий кристалічний осад. Таку саму
реакцію дають тартрати, а її проведенню заважають феноли і ароматичні
аміни, які утворюють осади з бромною водою:
1.2. Ідентифікація органічних лікарських речовин
Аналіз органічних лікарських речовин відрізняється від аналізу
неорганічних сполук. Молекула органічної речовини складається з основи
(скелету) (вуглеводневий (аліфатичний) ланцюг, ароматична або
гетероциклічна структура) і певного набору функціональних груп, наявність,
розташування та взаємний вплив яких і визначають хімічні й фармакологічні
C
CH2
CH2
COO
-
COO
-
COO
-
HO
2
Ca3
2+
3-
C
CH2
CH2
HO
COO
COO
COO
tº
+ 3Ca2+C
CH2
CH2
COO
-
COO
-
COO
-
2HO
+ 5Br2 2 C
CHBr2
CHBr3
OCO2 + 5HBr +C
CH2
CH2
COOH
COOH
O
+ CO2 + H2OC
CH2
CH2
COOH
COOH
O+ [O]C
CH2
CH2
HO
COOH
COOH
COOH
18.
19
властивості сполуки. Томуаналіз лікарських речовин органічної природи
зводиться до реакцій, направлених на визначення функціональних груп.
1.2.1. Ненасичені вуглеводні
Характерними реакціями ненасичених вуглеводнів є, головним чином,
реакції електрофільного приєднання:
1. При взаємодії лікарських речовин, які містять ненасичений зв’язок, з
бромною водою спостерігається знебарвлення останньої (наприклад, при
проведенні реакції талеохінної спроби на хінін).
Використання в такій реакції як середовища безводного розчинника,
наприклад, тетрахлорметану, дає змогу відрізнити алкени чи алкіни від
ароматичних сполук, які реагують з бромом з виділенням бромоводню (він не
розчиняється в тетрахлорметані й виділяється у вигляді бульбашок).
Методика. Розчин 1 г (0,3 мл) брому в 100 мл чотирихлористого вуглецю
додають краплями до розчину 50 мг речовини в 2 мл чотирихлористого
вуглецю. Розчин брому одразу знебарвлюється.
2. Калію перманганат в нейтральному або слаболужному середовищі
окиснює ненасичені сполуки, при цьому його розчини знебарвлюються:
В кислому середовищі, особливо при нагріванні, реакція не зупиняється
на стадії окиснення подвійного зв’язку і проходить далі з розривом вуглець-
вуглецевого зв’язку з утворенням відповідних альдегідів або, навіть, кислот:
Реакція не є специфічною, тому в фармацевтичному аналізі широкого
застосування не знаходить. Використовується вона, наприклад, для визначення
домішок цінаммоїлкокаїну й інших відновлюючих речовин у кокаїну
гідрохлориді.
В літературі описано використання цієї реакції для ідентифікації цинку
ундеценоату (а) та кислоти коричної (б).
Методика. а) 0,1 г цинку ундеценоату розчиняють у суміші 2 мл 1 моль/л
сульфатної кислоти і 5 мл льодяної оцтової кислоти (речовина не розчиняється
у воді) і додають по краплі 0,25 мл розчину калію перманганату –
спостерігається знебарвлення розчину переманганату калію;
б) 0,1 г коричної кислоти нагрівають з 0,1 г калію перманганату і 5 мл
1 моль/л сульфатної кислоти – утворюється бензальдегід, який визначають за
запахом.
3. π – Зв’язки ненасичених вуглеводнів можуть брати участь в утворенні
комплексів з солями важких металів, наприклад, стибію (III) хлоридом.
C C + [O] + H2O C C
OHOH
H
C C
OHO
+ [O] H2O + 2 C O
19.
20
Методика. 1 мгречовини розчиняють в 1 мл хлороформу; до отриманого
розчину додають 5 мл хлороформного розчину стибію хлориду – з’являється
відповідне забарвлення.
Ретинолу ацетат в таких умовах дає синє забарвлення, ергокальциферол
при взаємодії з хлороформним розчином стибію хлориду, що містить
2% ацетилхлориду, дає оранжево-рожеве забарвлення.
4. Характерними, однак недостатньо чутливими і неспецифічними
реакціями на ацетилен, його похідні є реакції з солями аргентуму, меркурію,
купруму (I), що призводять до утворення нерозчинних продуктів:
Cu2
2+
+ C2H2 + 2NH3 → C2Cu2↓ + 2NH4
+
В фармацевтичному аналізі деякі стероїдні гормони, що містять етинільні
радикали кількісно визначають методом непрямої алкаліметрії по нітратній
кислоті, що виділяється в результаті реакції з аргентуму нітратом.
1.2.2. Галоїдвмісні органічні сполуки
1. Найпростішою попередньою спробою на наявність галогену в складі
органічної речовини є спроба Бельштейна. Петельку на кінці тоненького
мідного дроту прожарюють поки вона перестане забарвлювати полум’я
спиртівки, ні до чого не торкаючись дають охолонути, набирають декілька
кристалів речовини і обережно, згори вниз, вносять у безбарвне полум’я. При
наявності галогену, полум’я забарвлюється у зелений колір.
Спроба Бельштейна не завжди дає правильний результат. Деякі речовини,
що містять азот і сірку, або здатні при нагріванні виділяти оксид вуглецю (II)
також викликають подібне забарвлення полум’я.
2. Органічні речовини, в яких галоген має ковалентний зв’язок з атомом
вуглецю в звичайних умовах не дають реакції з аргентуму нітратом. Деякі з
них, де цей зв’язок не дуже міцний, реагують при нагріванні з водно-спиртовим
розчином аргентуму нітрату (хлорбутин).
Щоб довести наявність галогену в молекулі органічної речовини, треба
перевести його в іоногенний стан. Наприклад, наважку хлорпропаміду
прожарюють у фарфоровому тиглі з сумішшю для спікання (суміш натрію і
калію карбонатів з калію нітратом), залишок розчиняють у гарячій воді,
фільтрують, підкислюють нітратною кислотою. Фільтрат дає реакцію на
хлориди.
За іншою методикою використовують “відновну мінералізацію”.
Методика. 0,1 г бромкамфори розчиняють у 3 мл спирту, додають 1 мл
розчину натрію гідроксиду, 0,3 г цинкового пилу і кип’ятять протягом 1-
2 хвилин. Суміш охолоджують, фільтрують; фільтрат дає характерні реакції на
броміди.
Дуже часто для переведення галогену в іоногенний стан використовують
реакцію лужного гідролізу при нагріванні з водним розчином натрію
гідроксиду (бромізовал, карбромал, циклофосфан, левоміцетин). Тривалість
нагрівання залежить від міцності зв’язку вуглець-галоген.
20.
21
3. Наявність атомівйоду визначають за появою фіолетових парів йоду під
час піролізу речовини (йодоформ) або окиснення її при нагріванні з розведеною
нітратною (хініофон) чи концентрованою сульфатною кислотою (білігност,
йодогност, сергозин).
4. Фторорганічні сполуки ідентифікують після мінералізації з сумішшю
для спікання. До розчину, що містить фторид-іони додають розчин кальцію
хлориду – з’являється біла каламуть (фторафур):
2F- + CaCl2 → CaF2↓ + 2Cl-
Більш сучасним є метод спалення у колбі з киснем. Продукти реакції
розчиняють у воді і додають до розчину феруму роданіду; спостерігається
значне послаблення забарвлення (фторурацил):
Fe(SCN)3 + 6F- → [FeF6]3- + 3SCN-
1.2.3. Спиртовий гідроксил
1. Найбільш загальною реакцією на спирти є реакція етерифікації.
Взаємодія спиртів з ангідридами, хлорангідридами або карбоновими кислотами
в присутності концентрованої сульфатної кислоти приводить до утворення
складних ефірів:
Продукти реакції ідентифікують за характерним запахом або
температурою плавлення.
Методика. а) 2 мл етанолу змішують з 0,5 мл льодяної оцтової кислоти, 1 мл
концентрованої сульфатної кислоти і нагрівають до кипіння – з’являється
характерний запах етилацетату;
б) до 0,1 г метиландростендіолу додають 0,6 мл оцтового ангідриду,
4,5 мл безводного піридину і нагрівають на водяному обігрівачі при 50-60°С
протягом трьох годин у колбі зі зворотним холодильником; до охолодженої
рідини додають 30 мл крижаної води. Через 30 хв осад відфільтровують,
переосаджують водою з ацетону, висушують і визначають температуру
плавлення отриманого моноацетату;
в) розчиняють 0,2 г ментолу в 0,5 мл безводного піридину, додають 3 мл
15% розчину хлорангідриду 3,5 динітробензойної кислоти в безводному
піридині і нагрівають на водяному обігрівачі протягом 10 хв; до отриманого
ROH +
RO H +
RO H +
O
C
C
R'
R'
O
O
C
O
R'RO + R'COOH
C R'
O
Cl C
O
R'RO + HCl
HO
O
R'C
H2SO4
C
O
R'RO + H2O
21.
22
розчину додають 7мл води і витримують у кризі протягом 30 хв. Осад
відфільтровують, промивають крижаною водою, перекристалізовують з
ацетону і визначають температуру плавлення.
2. Для ідентифікації спиртів широко застосовують реакції окиснення.
Утворені при цьому альдегіди або кетони встановлюють за характерним
запахом або відповідними якісними реакціями:
Ця реакція застосовується для підтвердження тотожності ефедрину
гідрохлориду, етанолу, бензилового спирту, а також для визначення домішки
метилового спирту в етиловому.
Методика. а) 0,05 г ефедрину гідрохлориду розчиняють в 1 мл води, додають
кристал калію фериціаніду і нагрівають до кипіння – з’являється запах
бензальдегіду:
б) до 0,1 мл бензилового спирту додають 5 мл 3% розчину калію
перманганату і 1 мл 1 моль/л сульфатної кислоти; утворюється бензальдегід,
який визначають за характерним запахом;
в) до 0,5 мл етанолу додають 4,5 мл води і 2 мл розчину калію
перманганату в фосфатній кислоті. Через 10 хв по краплі додають насичений
розчин натрію гідросульфіту до знебарвлення розчину, 1 мл 2% розчину
динатрієвої солі хромотропової кислоти, поступово, 10 мл концентрованої
сульфатної кислоти (порціями по 1 мл через 1 хв) і перемішують. Поява
фіолетового забарвлення свідчить про наявність домішки метилового спирту:
R CH2 OH
[O]
CR
H
O
CH
OH
R R'
[O]
CR R'
O
K3[Fe(CN)6]
C
H
O
. HCl
CH3NHOH
CH3CHCH
H2SO4 (конц.)
5CH3OH + 2KMnO4 + 3H3PO4 5H C
O
H
+ 2MnHPO4 + K2HPO4 + 8H2O
HO
+ H C
O
H
+
H2O
HO
SO3Na
SO3Na
NaO3S
OH
OH
NaO3S
22.
23
Однією з відомихреакцій на спирти, зокрема етанол, є реакція утворення
йодоформу:
Методика. До 0,5 мл етанолу додають 5 мл розчину натрію гідроксиду і 2 мл
0,1 моль/л розчину йоду – з’являється специфічний запах і поступово
утворюється жовтий осад йодоформу. Осад може здаватися аморфним, однак
під мікроскопом можна розпізнати гексагональні і зірчасті кристали.
Йодоформна реакція не є специфічною на спирти, оскільки сполуки, що
містять етоксильну (-OC2H5), ацетильну (-CO-CH3), моно і дийодацетильні
групи також дають позитивну спробу. В реакцію утворення йодоформу
вступають етанол, хлоралгідрат, оцтовий альдегід, метилкетони (наприклад,
ацетон) і вторинні спирти, які при окисненні утворюють метилкетони.
1.2.4. Багатоатомні спирти
1. Характерною властивістю багатоатомних спиртів є їх здатність
утворювати комплексні сполуки. Наприклад, при взаємодії з борною кислотою
утворюються комплексні кислоти і нейтральний або слаболужний, спочатку,
розчин стає кислим, що визначають за допомогою кислотно-основних
індикаторів:
Методика. 0,05 г речовини, що аналізується, розчиняють в 2 мл води, додають
краплю 0,02 моль/л розчину натрію гідроксиду і краплю розчину
фенолфталеїну. В іншій пробірці в 2 мл води розчиняють 0,5 г натрію
C
C
OH
OH
H
H
R
R'
+ H3BO32
C
C
H
H
R
R'
O
O
B
O
O C
C
R
R'
H
H
H
+
HO
[O]
HO
CHHO
SO3H
CH2
SO3H
HO3S
OH
OH
HO3S
SO3H
HO
SO3H
OH
HO3S
O
HO3S
R CH
OH
CH3 + NaOI
R C
O
CH3
+ NaI + H2OR C
O
CH3
+ 3NaOI R C
O
CI3 + 3NaOH
R C
O
CI3 + NaOH R C
O
ONa + CHI3
23.
24
тетраборату і додаютькраплю розчину фенолфталеїну. Обидва розчини
забарвлюються в рожевий колір, який зникає при їх змішуванні.
2. Розчини багатоатомних спиртів розчиняють купруму гідроксид з
утворенням сполук, забарвлених в інтенсивно-синій колір:
Методика. До 5 мл розчину купруму сульфату додають 1-2 мл розчину натрію
гідроксиду до утворення осаду купруму гідроксиду. Потім додають розчин
гліцерину до розчинення осаду. Розчин забарвлюється в інтенсивно-синій
колір.
3. Характерною для багатоатомних спиртів є також реакція дегідратації.
Наприклад, пентози в такій реакції утворюють фурфурол, а гліцерин – акролеїн:
Методика. Пробу, що містить гліцерин, змішують з подвійною кількістю калію
гідросульфату і нагрівають. Утворюється акролеїн, який ідентифікують за
характерним різким запахом, або за посинінням фільтрувального папірця,
змоченого свіжим 1% розчином натрію нітропрусиду, до якого додано краплю
піперидину.
Після лужного гідролізу таку реакцію дають складні ефіри гліцерину
(жири, нітрогліцерин). Цю реакцію дає також дипрофілін.
1.2.5. Енольний гідроксил
До енолів відносять сполуки, які знаходяться у рівновазі з відповідними
карбонільними сполуками:
Енольний гідроксил значно кисліший від спиртового і в цьому
відношенні близький до фенольного. За рахунок наявності енольного
гідроксилу етил- і фенілбарбітурова кислоти мають підвищену кислотність у
порівнянні з барбіталом і фенобарбіталом, їх розчини дають кислу реакцію за
метиловим червоним, що і використовується для відкриття домішки цих сполук
у відповідних субстанціях.
Наявність енольного гідроксилу зумовлює здатність сполук розчинятися
у лугах і утворювати забарвлені комплекси з солями важких металів.
CH2OH
CHOH
CH2OH
2 + Cu(OH)2
CH2
CH
CH2
O
OH
OH
Cu
HO
HO
O
CH2
CH
CH2
+ 2H2O
+ 2H2O
C
CH
CH2
H
O
KHSO4
HOH CH
CH OH
CH2 OH
R C CH
O
R1
R2
R C
OH
C
R1
R2
24.
25
Методика. 0,05 гбутадіону збовтують з 1,5 мл 0,1 моль/л розчину натрію
гідроксиду протягом 2 хв, фільтрують і до фільтрату додають 0,5 мл розчину
купруму сульфату – утворюється осад сіруватого кольору, який переходить у
блідо-блакитний:
1.2.6. Ендіольне угрупування
Характерним прикладом речовин, що належать до ендіолів є аскорбінова
кислота. Вона має кислі властивості і реагує з натрію гідроксидом:
1. Ендіоли, зокрема аскорбінова кислота, здатні утворювати комплекси з
солями важких металів.
Методика. До розчину 0,05 г аскорбінової кислоти в 2 мл води додають 0,1 г
натрію гідрокарбонату і 0,02 г феруму (II) сульфату, струшують і залишають
стояти – з’являється фіолетове забарвлення, яке зникає від додавання 5 мл
розведеної сульфатної кислоти.
2. Ендіоли називають ще редуктонами, оскільки вони характеризуються
високою відновною здатністю і легко дегідруються з утворенням відповідних
α-дикарбонільних сполук. При цьому вони в нейтральному або слабокислому
середовищі відновлюють аргентуму нітрат до металічного срібла, солі купруму
(II) до сполук купруму (I), знебарвлюють розчин йоду, відновлюють хіноїдні
барвники до лейкосполук. Наприклад: при додаванні до розчину аскорбінової
кислоти (1:1000) по краплі розчину 2,6-дихлорфеноліндофенолу синє
забарвлення зникає.
O
OH
O
NaO
+ NaOH
O
OH
O
HO
HOCH
CH2OH
HOCH
CH2OH
2
N
N
OHH9C4
O C6H5
C6H5
NaOH
H2O
N
N
H9C4
O C6H5
C6H5
ONa
CuSO4
N
N
H9C4
O C6H5
C6H5
O
-
Cu
2+
N
N
H9C4
O C6H5
C6H5
O
25.
26
1.2.7. Загальні якісніреакції фенолів
До фенолів відносять сполуки в яких гідроксильна група приєднана до
ароматичного радикалу.
1. Однією з характерних властивостей фенолів є їх слабка кислотність.
Феноли розчиняються в 5% розчині натрію гідроксиду з утворенням фенолятів і
не розчиняються у розчині натрію карбонату.
Методика. До 5 мл 2% розчину морфіну гідрохлориду додають 1 краплю
розчину амоніаку – утворюється білий кристалічний осад, який розчиняється
при додаванні надлишку розчину натрію гідроксиду.
Кислотні властивості фенолів підсилюються в присутності
електроноакцепторних замісників і такі сполуки набувають розчинності в
розчинах карбонатів (трихлорфенол, нітрофеноли, 8-оксихінолін).
Методика. До 1 мл 1% розчину хінозолу додають розчин натрію карбонату –
утворюється осад, який розчиняється в надлишку реактиву:
В залежності від наявності в молекулі інших функціональних груп деякі
феноли розчиняються в розчинах натрію гідроксиду з утворенням забарвлених
розчинів. Наприклад, рутин дає жовте забарвлення, нітроксолін – оранжево-
червоне, дантрон – червоне.
Методика. 0,05 г фтивазиду розчиняють при слабкому нагрівання в 10 мл 95%
спирту і охолоджують. Від додавання однієї краплі розчину натрію гідроксиду
світло-жовте забарвлення розчину переходить в оранжево-жовте.
OH
Cl
OO
OH OH
H
CH
CH2OH
HO
+
OO
O
H
CH
CH2OH
HO
O
+
OH
N
O
Cl
OH
Cl
NH
Cl
2
2
Na2CO3. H2SO4 + Na2CO3 + Na2SO4 + CO2 + H2O
N
OH
N
OH
+ NaHCO3
N
ONa
26.
27
2. Найбільш характерноюреакцією на сполуки, що мають фенольний
гідроксил є реакція з феруму (III) хлоридом. Реакція притаманна лише самим
фенолам; ні прості, ні складні ефіри фенолів без гідролізу її не дають.
Рекомендується застосовувати нейтральні або у крайньому разі слабокислі
водні розчини. Феноли, в яких в орто-положенні до групи –OH відсутні групи
здатні утворювати комплекси, дають забарвлення тільки у водному розчині,
причому воно поступово блідне і зникає при додаванні спирту, мінеральних
кислот і солей амонію. Якщо ж в орто-положенні до фенольного гідроксилу є
групи здатні утворювати комплекси (альдегідна, кетонна, карбоксильна,
гідрокси-, алкокси- або карбалкоксигрупи), то забарвлення, внаслідок
утворення хелатованих солей, виникає як в водному, так і в спиртовому
розчині. Додецилгалат дає забарвлення з розчином FeCl3 в водно-ацетоновому
розчині.
Характер забарвлення при взаємодії фенолів з розчином феруму (III)
хлориду залежить від наявності в ароматичному ядрі інших функціональних
груп, їх розташування, а також кількості і розташування фенольних гідроксилів
у ядрі.
Як правило готують 1% водний розчин аналізованої речовини; до 2-3 мл
розчину додають краплю 0,5% розчину феруму (III) хлориду. Забарвлення
з’являється зразу й іноді зберігається довго, а іноді зникає через 1-2 хв. Якщо
сполука не розчиняється у воді можна розчинити її в етанолі й розбавити
розчин водою. В результаті утворюється тонкодиспергований у воді фенол,
який дає позитивну реакцію.
Фенол, саліцилова кислота, саліциламід дають з розчином феруму (III)
хлориду фіолетове забарвлення; резорцин – синьо-фіолетове; рутин
діетилстільбестрол, хініофон, адреналін – зелене; фтивазид – жовтувато-зелене;
піридоксину гідрохлорид – червоне; хлортетрацикліну гідрохлорид –
коричневе, що переходить у червоне.
3. Одним з характерних реактивів, які використовують для ідентифікації
фенолів і їх похідних, зокрема фенолкарбонових кислот, є реактив Мілона –
розчин меркурію в нітратній кислоті. Феноли дають з ним червоне забарвлення.
Іноді, як наприклад, для самого фенолу реакція є дуже чутливою.
Методика. 0,1 г бензилгідроксибензоату (бензиловий ефір 4-гідроксибензойної
кислоти) розчиняють у 2 мл 96% етанолу і додають 0,5 мл розчину меркурію у
нітратній кислоті – утворюється осад і поступово рідина над ним забарвлюється
в червоний колір (BP).
4. Так само як і спирти, феноли можуть вступати в реакції етерифікації з
ангідридами, галоїдангідридами карбонових кислот:
Продукти реакції ідентифікують за температурою плавлення.
O C R
O
R'OHR'
(RCO)2O ; RCOCl
27.
28
Методика. До 0,25г синестролу додають 1 мл оцтового ангідриду і 2мл
безводного піридину. Кип’ятять зі зворотним холодильником протягом 15 хв,
охолоджують, додають 50 мл води і ретельно струшують. Осад
відфільтровують, промивають водою, сушать при 100-105оС. Температура
плавлення виділеного діацетату синестролу складає 137-139оС.
5. Особливістю фенолів у порівнянні зі спиртами є здатність досить легко
окиснюватися під дією різноманітних окисників. В результаті утворюються
продукти, які мають характерне забарвлення.
При змочуванні декількох крупинок апоморфіну гідрохлориду 1 краплею
нітратної кислоти з’являється криваво-червоне забарвлення.
6. Хінони, які утворюються при окисленні фенолів конденсуються з
амінами з утворенням хінонімінів і продуктів їх конденсації. Прикладом такої
реакції є реакція індофенолової спроби.
Методика. 0,05 г речовини, що досліджується, розчиняють в 0,5 мл розчину
амоніаку і додають 3-4 краплі розчину хлораміну (хлорного вапна, натрію
гіпохлориту, бромної води) суміш нагрівають на водяному обігрівачі – через
декілька хвилин з’являється забарвлення, яке змінюється від додавання
кислоти:
Реакцію окиснення в присутності амоніаку дають не лише феноли але й
деякі їх прості ефіри. Прикладом може слугувати реакція талеохінної спроби.
Методика. До 5 мл 0,1% розчину хініну гідрохлориду додають 2-3 краплі
бромної води і 1 мл розчину амоніаку – з’являється зелене забарвлення:
індофенол (синє забарвлення)
H C6H4OH
H2O
NH3[O]
O
O
O
NH
O
N OHOH
N
CH CH2
Br2
N
CH CH2
OH OH
N
CH CH2
Br Br
4NH4OH
N
H3CO
CH
OH
N
N
O
O CH
OH
CH
OHNH
NH
28.
29
Різновидом реакції індофеноловоїспроби є нітрозореакція Лібермана,
характерна для фенолів, які не мають замісників в о- і п-положеннях. Під дією
нітритної кислоти утворюється п-нітрозофенол, який ізомеризується в п-
хіноїдоксим, що реагуючи з надлишком фенолу в кислому середовищі дає
забарвлений індофенол:
Методика. Крупинку речовини вміщують на фарфорову пластинку або
годинникове скло, змочують 2-3 краплями 1% розчину натрію нітриту в
концентрованій сульфатній кислоті – спостерігається забарвлення, яке
змінюється від додавання розчину лугу.
7. Фенольний гідроксил є потужним орієнтантом першого роду і тому
велику групу якісних реакцій фенолів складають реакції ароматичного ядра –
реакції конденсації та реакції електрофільного заміщення. Розчини фенолів при
взаємодії з бромною водою утворюють осади білого кольору:
Ця реакція використовується для кількісного визначення фенолів
методом броматометрії.
8. Феноли, які мають незаміщене пара- або орто-положення легко
взаємодіють з солями діазонію з утворенням азобарвників. При додаванні солі
діазонію до лужного розчину фенолу утворюється інтенсивно забарвлений
розчин або осад:
Методика. 0,05 г речовини розчиняють у 5 мл води, додають 2 мл розчину
амоніаку і 1 мл діазореактиву – спостерігається забарвлення, зумовлене
утворенням відповідного азобарвника.
9. Для фенолів, у яких вільне п-положення характерною є реакція
конденсації з 2,6-дихлорхінонхлорімідом:
+
HCl
NaNO2
індофенол
п-хіноїдоксимп-нітрозофенол
HO HO NO O NOH
HO H HON O HO N O
+ 3HBr+ 3Br2
OH
BrBr
OH
Br
OHN NN NONH4 HO3S HO3S+
+
Cl
-
_
NH4Cl
29.
30
Методика. 0,01 гпіридоксину гідрохлориду розчиняють в 10 мл води; до
0,1 мл отриманого розчину додають 1 мл води, 2 мл амоніачного буферного
розчину, 1 мл розчину 2,6-дихлорхінонхлоріміду, 2 мл бутилового спирту і
струшують протягом 1 хв – шар бутилового спирту забарвлюється в блакитний
колір.
10. Феноли та їх прості ефіри здатні вступати в реакцію конденсації з
формальдегідом в присутності концентрованої сульфатної кислоти (реактив
Маркі) з утворенням забарвлених арилметинових барвників:
Методика. До 0,03 г саліцилової кислоти додають 5 мл концентрованої
сульфатної кислоти і 2 краплі формаліну. Суміш підігрівають – з’являється
червоне забарвлення. Або: декілька крупинок досліджуваної речовини
вміщують на годинникове чи предметне скло, змочують 2-3 краплями реактиву
Маркі. При стоянні або після легенького підігрівання спостерігається
відповідне забарвлення.
1.2.8. Альдегіди і кетони
Структурною особливістю альдегідів і кетонів є наявність карбонільної
групи >C=O. Для альдегідів і кетонів характерні 4 групи реакцій:
1. Реакції окиснення (альдегіди).
2. Реакції приєднання.
3. Реакції полімеризації і конденсації.
4. Реакції заміщення.
Альдегіди
1. Окисно-відновні реакції. Найбільш розповсюдженими реакціями
альдегідів є реакції відновлення іонів металів (аргентуму, купруму, меркурію).
Такі реакції дозволяють відрізнити альдегіди від кетонів.
N
CH2OH
CH2OHHO
H3C
NCl O
Cl
Cl O
Cl
Cl
+
HCl
N
CH2OH
CH2OHHO
H3C N
H2SO4+C
O
H H+
HOOC H
HO
H
OH
COOH
HOOC
HO
CH2
OH
COOH HOOC
HO
CH
O
COOH
30.
31
а) Реакція “срібногодзеркала”. Реактивом є амоніачний розчин оксиду
аргентуму (реактив Тойленса). В результаті реакції альдегід окислюється до
кислоти, а окис аргентуму відновлюється до металічного срібла:
Методика. В добре вимиту пробірку наливають розчин аргентуму нітрату і
краплями додають розчин амоніаку до розчинення осаду, що утворився
спочатку. Приливають 2-3 краплі розчину альдегіду і обережно нагрівають на
водяному обігрівачі – виділяється металічне срібло у вигляді дзеркала чи сірого
осаду.
б) Реакція з реактивом Фелінга.
Методика. До розчину 0,2 г глюкози в 5 мл води додають 10 мл реактиву
Фелінга і нагрівають до кипіння – випадає цеглинно-червоний осад:
в) Реакція з реактивом Несслера. Альдегіди відновлюють реактив
Несслера з утворенням осаду металічної ртуті:
Реакція з реактивом Несслера є більш чутливою, ніж реакції з реактивами
Тойленса і Фелінга, і тому її застосовують для відкриття домішок альдегідів в
інших речовинах, наприклад, у діетиловому ефірі.
Разом з тим треба пам’ятати, що ці реакції не є специфічними, їх дають
будь які відновники – ендіоли, о-, п-дифеноли, похідні гідразину і т. ін.
Альдегіди і кетони
1. Реакції приєднання. Серед реакцій приєднання слід зупинитися на
реакціях з бісульфітом натрію. Альдегіди і кетони реагують з ним і дають
бісульфітні похідні, які добре кристалізуються:
C
O
H
R + 2[Ag(NH3)2]OH C
O
R
ONH4
+ 2Ag + 3NH3 + H2O
C
O
H
R + K2HgI4 + 3KOH R COOK + 4KI + Hg + 2H2O
C
O
H
R + NaHSO3 C
H
R
OH
SO3Na
HOHC
HOHC
NaOOC
OOC
CHOH
CHOH
COOK
COO
Cu
C
C
C
C
C
CH2OH
OH
OH
OH
HO
H
H
H
H
H
O
2+ + 4
2CuOH
t Cu2O + H2O
3
COONa
C
C
COOK
H
H
OH
OH
NaOH
C
C
C
C
CH2OH
OH
OH
OH
H
H
H
HO
H
COONa
2KOH
+2CuOH +2H2O
31.
32
З утворених бісульфітнихпохідних альдегід або кетон можна виділити
шляхом лужного чи кислотного гідролізу. Ця реакція застосовується для
очистки альдегідів і кетонів, а також для виділення їх з сумішей речовин,
наприклад, ефірних олій.
З аналітичною метою частіше застосовують реакцію з фуксинсульфітною
кислотою. Фуксинсульфітна кислота – безбарвна речовина, яка при взаємодії з
розчинами альдегідів або деяких кетонів поступово дає червоно-фіолетове
забарвлення:
Для більшості сполук, крім формальдегіду, забарвлення зникає від
додавання мінеральної кислоти. Глюкоза такої реакції не дає через низьку
концентрацію у водному розчині альдегідної форми.
2. Реакції конденсації. Реакцію конденсації формальдегіду з фенолами ми
вже розглядали на прикладі саліцилової кислоти (див. феноли, 1.2.7.10). В
фармацевтичному аналізі її використовують для відкриття сполук, які можуть
утворити формальдегід у процесі окиснення (метанол) або гідролізу
(гексамідин, метазид, нікодин).
Аліфатичні та ароматичні альдегіди вступають в реакцію конденсації з
первинними ароматичними амінами. В експрес-аналізі застосовується реакція з
альдегідами лігніну (див. первинні ароматичні аміни, 1.2.13.4).
3. Реакції заміщення. Кетони, як і альдегіди, при взаємодії з
гідроксиламіном, фенілгідразином, 2,4-динітрофенілгідразином, семікарбази-
дом дають реакцію конденсації, яку прийнято називати реакцією заміщення –
заміщення карбонільної групи на залишок відповідного реактиву.
Методика. а) 0,05 г тестостерону пропіонату в колбі зі зворотним
холодильником нагрівають на водяному обігрівачі протягом 1 години з 7 мл
реактиву (0,05 г гідроксиламіну гідрохлориду, 0,05 г натрію ацетату в 25 мл
спирту). До охолодженої рідини додають 15 мл води. Осад, що випав,
відфільтровують, промивають водою, перекристалізовують з 50% спирту,
сушать і визначають температуру плавлення (166-171оС):
C
H2SO3
R CHO
NH SO2H
CHO3S NH2
NH SO2H NH SO2 C
H
OH
R
CHO3S NH2
NH SO2 C
H
OH
R
NH
NH SO2 C
H
OH
R
NH SO2 C
H
OH
R
CH3
H
H
O C CH2 CH3
O
O
CH3 H
H
NH2OH . HCl
CH3COONa
CH3
H
H
O C CH2 CH3
O
CH3 H
H
HON
32.
33
б) 1 мгпреднізолону розчиняють в 1 мл метилового спирту, додають 5 мл
розчину сульфату фенілгідразину і нагрівають на водяному обігрівачі – через
5 хв з’являється жовте забарвлення:
в) 0,02 текодину розчиняють у 1 мл води і додають 2 мл розчину 2,4-
динітрофенілгідразину в хлороводневій кислоті – при струшуванні швидко
випадає осад жовтого кольору:
Ці реакції застосовуються для ідентифікації карбонільних сполук, а також
для їх кількісного визначення методом непрямої алкаліметрії, гравіметрії,
фотоелектроколориметрії.
1.2.9. Карбонові кислоти
Карбонові кислоти – органічні сполуки, які містять карбоксильну
групу –COOH.
1. Утворення складних ефірів при взаємодії зі спиртами. Якщо тотожність
спиртів підтверджують по реакції утворення складних ефірів з карбоновими
кислотами, то так само карбонові кислоти можна ідентифікувати по утворенню
зі спиртами складних ефірів, що мають характерний запах. Наприклад, при
нагріванні саліцилової кислоти або її солі з метанолом в присутності
концентрованої сульфатної кислоти виникає характерний запах
метилсаліцилату:
+ H2SO4 + 2H2O2
2
CH3
H
OH
C CH2
O
OH
+
2
. H2SO4
NHNC6H5
C CH2
O
OHCH3
H
OH
O
HO
H
HCH3
CH3 H
H
HO
NHNH2
N
OH
O
O
H3CO
CH3
. HCl + +. HCl
CH3
O
H3CO
N
OH H2O
NO2
NO2
NHNH2
O2N
NO2
NHN
33.
34
2. Карбонові кислотимають значно вищу кислотність в порівнянні зі
спиртами і фенолами. Ідентифікують їх, як правило, по реакції утворення
різноманітних солей. В залежності від структури молекули і наявності інших
функціональних груп різні кислоти дають різноманітні забарвлені або
нерозчинні у воді солі з різними металами. Наприклад, винна кислота утворює
білий осад калію гідротартрату; альгінова кислота утворює драглисті
желатиноподібні осади з солями кальцію та магнію; цитринова кислота
утворює кальцієву сіль, яка розчиняється у воді на холоду і не розчиняється при
кипінні:
Найчастіше для підтвердження тотожності карбонових кислот та їх солей
використовують реакції з солями Cu2+
, Fe3+
, Co2+
, дещо рідше з солями Ag+
та
Pb2+
. Найбільш загальною методикою ідентифікації карбонових кислот є така:
невелику кількість досліджуваної речовини нейтралізують 0,1М розчином
натрію гідроксиду (по фенолфталеїну) до блідо-рожевого забарвлення.
Отриманий розчин розливають у пробірки, в одну з яких додають розчин
феруму (III) хлориду, в другу – розчин кобальту (II) хлориду (або нітрату) і в
третю – купруму (II) сульфату. В усіх пробірках в залежності від конкретної
кислоти спостерігається відповідне забарвлення або осад (див. аніони, 1.1.2).
3. Карбонові кислоти при нагріванні до температури 150-160оС
декарбоксилюються, при цьому деякі з них утворюють речовини, що мають
характерний запах.
Методика. а) 0,1 г нікотинової кислоти нагрівають з 0,1г безводного натрію
карбонату – з'являється запах піридину:
б) 0,1 г саліцилової кислоти нагрівають з 0,3 г цитрату натрію –
відчувається запах фенолу:
+ 6NaClCa3
2
CHO CH2COO
-
CH2COO
-
CH2COO
-
tº
C COONa2HO
CH2
CH2
COONa
COONa
+ 3CaCl2
+ CH3OH
H2SO4
+ H2O
OH
COOH
OH
COOCH3
+ CO2
Na2CO3
tº
N N
COOH
tº + CO2
COOH
OH
OH
34.
35
в) 1 гсаліцилової кислоти нагрівають з 2 мл концентрованої сульфатної
кислоти і газ, що виділяється, пропускають через вапняну воду – з’являється
каламуть: CO2 + Ca(OH)2 → CaCO3↓ + H2O
Карбонат і цитрат натрію додають аби втримати речовину від сублімації
до досягання температури, необхідної для декарбоксилювання.
1.2.10. Амінокислоти аліфатичного ряду
1. Найбільш спільною реакцією амінокислот є реакція утворення
комплексів з солями купруму (II). До нейтрального або слаболужного розчину
амінокислоти додають декілька крапель сульфату або ацетату купруму –
з’являється інтенсивно-синє або синьо-фіолетове забарвлення:
2. Груповою реакцією на амінокислоти є реакція з нінгідрином.
Методика. До 0,1-0,2% водного розчину амінокислоти додають декілька
крапель розчину нінгідрину і нагрівають до кипіння – утворюються продукти
конденсації синього або синьо-фіолетового кольору:
Необхідно враховувати, що нінгідрин утворює забарвлені продукти
реакції не лише з амінокислотами, але й з первинними амінами, гідразидами
кислот і деякими іншими речовинами.
+ Na2SO4 + 2H2O
O
CH
NH2
C O
Cu
NH2
O
CO
RCH
R
2NaOH
+ CuSO42R CH COOH
NH2
C
CO
C
O
O
. H2O + R CH COOH
NH2
+ R C
O
H
+ NH3 + CO2
C
C
O
O
CHOH
NH3
сіль синьо-фіолетового кольру
O + H2 N H + HO H
C
C
O
O
C
C
C
O
O
C
C
C
O
O
C N
O
OH
C
C
C
C
O
O
C N
O
C
C
ONH4
35.
36
1.2.11. Прості ефіри
Простіефіри – це оксигенвмісні органічні сполуки загальної формули:
R–O–R1
1. Найбільш простим способом ідентифікації простих ефірів в органічній
хімії є перетворення їх в леткі алкілйодиди при нагріванні з концентрованою
йодоводневою кислотою:
R–OCH3 + HI → ROH + CH3I
R – OC2H5 + HI → ROH + C2H5I
Виділені алкілйодиди ідентифікують за температурою кипіння. Метод
неспецифічний, оскільки відповідну реакцію дають також спирти.
2. Наявність двох неподілених пар електронів на атомі оксигену надає
простим ефірам дуже слабких основних властивостей, однак у середовищі
концентрованої сульфатної кислоти вони можуть утворювати оксонієві солі.
Методика. На годинникове скло наносять 3-4 краплі концентрованої
сульфатної кислоти і додають 0,02 г димедролу – з'являється яскраво-жовте
забарвлення, що поступово переходить у цеглинно-червоне. Від додавання
декількох крапель води забарвлення зникає:
Схожу реакцію дає і структурний аналог димедролу – дифенілпіраліну
гідрохлорид.
1.2.12. Складні ефіри
Складні ефіри – це сполуки загальної формули:
1. Найбільш важливою властивістю складних ефірів, з аналітичної точки
зору, є їх здатність піддаватися гідролізу, який каталізується кислотами або
основами з утворенням відповідного спирту і кислоти (або її солі).
Методика. а) 0,2 г ацетилсаліцилової кислоти вміщують у фарфорову чашку,
додають 0,5 мл концентрованої сульфатної кислоти, перемішують і додають 1-2
краплі води – відчувається запах оцтової кислоти:
б) до 0,02 г фенілсаліцилату додають 3-4 краплі концентрованої
сульфатної кислоти і 1-2 краплі води – відчувається запах фенолу:
R C
O
O R'
H2SO4
H2O
+ CH3COOH
O C CH3
O
COOH COOH
OH
H2SO4
H2O
+
OH
C
O
O
OH
OH
COOH
++
H
H
CH3
CH3
NCH
C6H5
C6H5
CH2OCH SO4
2- + HCl
H2SO4 конц.. HClCH O CH2 CH2 N
CH3
CH3
C6H5
C6H5
2
36.
37
2. Під часгідролізу складних ефірів у лужному середовищі в присутності
гідроксиламіну утворюються гідроксамові кислоти, які з важкими металами
дають забарвлені солі – гідроксамати. Найчастіше використовують
гідроксамати феруму (III), які в залежності від складу солі забарвлені в
червоно-бурий, вишнево-червоний або червоно-фіолетовий колір.
Методика. 1 мг кортизону ацетату розчиняють у 2 мл метилового спирту і
додають 2 мл лужного розчину гідроксиламіну. Струшують протягом 3-5 хв,
додають 2 мл розведеної хлороводневої кислоти і 0,5 мл 10% феруму (III)
хлориду в 0,1 моль/л розчині хлороводневої кислоти – з’являється темно-
вишневе забарвлення:
Окрім складних ефірів аналогічну реакцію дають лактони, а також β-лактами
(лікарські речовини групи пеніциліну).
3. Окремим випадком складних ефірів можна вважати лактони –
внутрішньомолекулярні циклічні складні ефіри. Специфічною реакцією на
п’ятичленне лактонне кільце є реакція Легаля.
Методика. До розчину 1-2 мг целаніду в 1 мл 95% спирту додають 1 мл
розчину натрію нітропрусиду і 1-2 краплі розчину натрію гідроксиду –
з’являється і поступово зникає червоне забарвлення.
Найчастіше цю реакцію застосовують для підтвердження тотожності
серцевих глікозидів з групи карденолідів, але її дає також алкалоїд, який має
п’ятичленне лактонне кільце, – пілокарпін.
1.2.13. Аміни
Аміни – це похідні амоніаку, в яких 1, 2 або 3 атоми гідрогену заміщені
на алкільні або арильні радикали. Відповідно розрізняють аліфатичні й
ароматичні аміни, первинні, вторинні, третинні аміни. Аміни є основами, що
зумовлено наявністю в атому азоту неподіленої електронної пари. Приєднуючи
протон, вони утворюють солі. Основність залежить від природи і числа
радикалів при азоті. Первинні аміни R–NH2 менш основні, ніж вторинні
R–NH–R'. У третинних амінів основність знижена за рахунок просторових
утруднень. Зменшення основності ароматичних амінів у порівнянні з
аліфатичними пояснюється спряженням неподіленої пари електронів азоту з
електронами ароматичного ядра.
Для ідентифікації амінів використовують різноманітні реакції. Аміни
визначають попередньою спробою, що ґрунтується на їх здатності утворювати
H
+
; FeCl3
3
_CH3 C
NHO
O
CH3 C
O
NHONa
Fe
3+
C
O
CH2 O C
O
CH3
OH
NH2OH
NaOH
C
O
CH2OH
OH
+
CH3 C
O
NHONa
3 + 3NaCl
37.
38
солі й наїх розчинності. Сполуки, розчинні в діетиловому ефірі і нерозчинні у
воді, але розчинні в 5% розчині хлороводневої кислоти можуть бути амінами.
1. Первинні й вторинні аміни дають реакцію ацилювання – заміни
гідрогену в групах –NH2 і –NH– на ацильний радикал. Ацилюють найчастіше
оцтовим ангідридом, ацетилхлоридом або бензоїлхлоридом. Продукти реакції
ідентифікують за характерною температурою плавлення. Третинні аміни не
ацилюються.
Методика. 1 г фенаміну розчиняють в 50 мл води, додають 10 мл розчину
натрію гідроксиду, 0,5 мл бензоїлхлориду і струшують; повторюють додавання
бензоїлхлориду по 0,5 мл до припинення утворення осаду. Температура
плавлення осаду після подвійної перекристалізації з 50% етанолу – 132-135оС.
2. Неподілена пара електронів не тільки надає амінам основних
властивостей, але й може вступати в реакцію комплексоутворення. Так само як
і амінокислоти, аміни в лужному середовищі утворюють комплекси з солями
купруму (II).
Методика. а) 0,1 г еуфіліну розчиняють в 4 мл води. До 3 мл цього розчину
додають 5 крапель розчину купруму сульфату – з’являється яскраве фіолетове
забарвлення:
б) 0,01 г ефедрину гідрохлориду розчиняють в 1 мл води, додають 0,1 мл
розчину купруму сульфату й 1 мл розчину натрію гідроксиду – з’являється синє
забарвлення. При збовтуванні цього розчину з 1 мл ефіру ефірний шар
забарвлюється в фіолетово-червоний колір, а водний зберігає синє забарвлення:
SO4
CH2
CH2
NH2
NH2
CuCuSO4+2
CH2
CH2
NH2
NH2 2
2
. H2SO4 + 2 + 4NaOH 2
+ 2NaCl + Na2SO4 + 2H2O
CH2
CH
NH2
CH3
C
O
Cl
CH2
CHCH3
NH C
O
+ 2H2O
+ Na2SO4 + 2NaCl +. HCl + CuSO4 + 2NaOH2
NHCH3OH
CH3
CH3
CH3
H H
CC
CH
O
Cu
O
CH CH
NHCH3
NHCH3
CH
38.
39
3. Первинні аліфатичній ароматичні аміни, а також вторинні аміни
можна розрізнити за допомогою реакції з нітритною кислотою. Первинні аміни
в реакції з нітритною кислотою утворюють діазосполуки. Але, якщо ароматичні
діазосполуки стійкі, то аліфатичні – нестабільні і одразу після утворення, навіть
на холоду, швидко розкладаються з виділенням азоту:
R–NH2 + ONOH → R–OH + H2O + N2↑
Загальна методика виглядає так: в мікропробірці змішують декілька
міліграмів натрію нітриту з хлороводневокислим розчином аміну. Інтенсивне
виділення безбарвного газу – азоту свідчить про наявність у сполуки первинної
аліфатичної аміногрупи.
В кислому середовищі сам нітрит натрію розкладається з утворенням
окислів азоту. При уважному розгляданні видно, що над шаром рідини
збирається бурий газ:
NaNO2 + HCl → NaCl + HNO2
2HNO2 → H2O + NO↑ + NO2↑
Методика. Розчиняють 0,1 г мексилену гідрохлориду [(RS)-1-метил-2-(2,6-
ксилілокси)-етиламіну гідрохлориду] в 3 мл 0,02 моль/л хлороводневої кислоти
і додають декілька кристалів натрію нітриту – спостерігається бурхливе
виділення газу.
Вторинні аміни при взаємодії з нітритною кислотою утворюють
нітрозаміни:
Продуктом реакції найчастіше є жовта важка рідина або кристалічний
осад, розчинні в діетиловому ефірі. Ефірний шар відділяють і випаровують до
сухого залишка. Отриманий нітрозамін при нагріванні з концентрованою
сульфатною кислотою гідролізується з утворенням нітритної кислоти, яка в
присутності фенолу дає реакцію Лібермана (див. феноли, 1.2.7.6).
Первинні ароматичні аміни при взаємодії в кислому середовищі з
азотистою кислотою утворюють безбарвні або блідо-жовті солі діазонію, які з
лужними розчинами фенолів дають реакцію азосполучення з утворенням
азобарвників.
Методика. 0,05 г анестезину розчиняють в 2 мл води, підкисленої 3 краплями
розведеної хлороводневої кислоти, додають 3 краплі 0,1 моль/л розчину натрію
нітриту і збовтують; отриманий розчин додають до 3 мл лужного розчину β-
нафтолу – з’являється вишнево-червоне забарвлення або утворюється
оранжево-червоний осад:
NH + HONO
R
R1
N NO
R
R1
+ H2O
39.
40
При проведенні реакціїдіазотування співвідношення реагентів має
становити мінімум 2,5 моля хлороводневої кислоти на 1 моль ароматичного
аміну. З яких 1 моль витрачається на утворення нітритної кислоти в реакції з
натрію нітритом, 1 моль – на утворення солі діазонію, а мінімум 0,5 моля – на
створення кислого середовища, в якому сіль діазонію є найбільш стійкою. При
проведенні реакції азосполучення сіль діазонію додають до лужного розчину β-
нафтолу оскільки в кислому середовищі феноли, зокрема β-нафтол не
розчиняються, випадають в осад, який може не реагувати з солями діазонію.
Поява жовто-буро-зеленого осаду свідчить про негативний результат реакції
азосполучення. Це в кислому середовищі випав осад β-нафтолу і продуктів його
окиснення нітритною кислотою.
Деякі солі діазонію мають забарвлення і не потребують проведення
реакції азосполучення.
Методика. 0,05 г етакридину лактату розчиняють в 5 мл води, підкислюють
розведеною хлороводневою кислотою і додають 1 мл розчину натрію нітриту –
з’являється вишнево-червоне забарвлення.
На реакції утворення солей діазонію ґрунтується нітритометричний метод
кількісного визначення ароматичних амінів.
4. Первинні ароматичні аміни при взаємодії в кислому середовищі з
аліфатичними або ароматичними альдегідами (п-диметиламінобензальдегід,
альдегіди лігніну, ванілін і т. ін.) утворюють забарвлені в жовтий або
оранжевий колір основи Шиффа:
NaOH
HO
+Cl
-
+
2,5 HCl
NaNO2
азобарвник вишнево-червоного
кольору
NH2
C
O
OR
C
O
OR
N N
NaO
N N
C
O
OR
N
NaNO2
+
HCl
+ CHOH C
O
CH3 OH
. CH3 CHOH C OH
O
Cl
-
NNN
NH2
OC2H5
H2N
NH2
OC2H5
40.
41
Методика. До 1мл 0,1% розчину норадреналіну гідротартрату додають 1 мл
1% розчину 2,5-діетокситетрагідрофурану в льодяній оцтовій кислоті.
Нагрівають до 80о
С протягом 2 хвилин охолоджують на льоду і додають 3 мл
2% розчину 4-диметиламінобензальдегіду в суміші 1 об’єму хлороводневої і 19
об’ємів льодяної оцтової кислоти; змішують і витримують 2 хвилини –
утворюється інтенсивне рожеве забарвлення.
В експрес-аналізі спробу на первинну ароматичну аміногрупу проводять
за такою методикою: кристалик речовини, що аналізується, вміщують на
клаптик небіленого газетного паперу і змочують краплею розведеної
хлороводневої кислоти – утворюється пляма оранжевого кольору.
5. Первинні ароматичні аміни при нагріванні зі спиртовим розчином лугу
і хлороформом утворюють ізонітрили, які мають неприємний нудотний запах:
6. Так само як і фенольний гідроксил, аміногрупа є орієнтантом першого
роду і тому ароматичним амінам притаманні реакції електрофільного
заміщення. Розчин аніліну при взаємодії з бромною водою дає білий осад 2,4,6-
триброманіліну:
Подібні реакції лежать в основі кількісного визначення багатьох
ароматичних амінів методом броматометрії.
7. Ароматичні аміни при взаємодії з концентрованою нітратною
кислотою легко нітруються. Утвореним нітропохідним за рахунок наявності
рухомого гідрогену аміногрупи притаманне явище нітро-ацинітротаутомерії,
тобто вони дають реакцію подібну до Віталі-Морена.
Методика. 0,01 г дикаїну вміщують в фарфорову чашку, змочують 2-3
краплями концентрованої нітратної кислоти і випарюють на водяному
обігрівачі насухо. До охолодженого залишку додають декілька крапель
0,5 моль/л спиртового розчину гідроксиду калію – з’являється криваво-червоне
забарвлення:
NH2 + CHCl3 + 3NaOHR R N C + 3NaCl + 3H2O
+ H2O+R NH2 C
O
H
N
CH3
CH3
R N CH
CH3
CH3
N
Br
NH2
Br
Br
NH2
+ 3Br2
+ 3HBr
41.
42
Реакція не єспецифічною для вторинних амінів, в літературі описано її
використання для підтвердження тотожності прокаїну гідрохлориду
(новокаїну).
8. Для виявлення третинних амінів рекомендується реакція з цитриновою
кислотою. Розчини цитринової, аконітової, малонової кислот в оцтовому
ангідриді при нагріванні з третинними амінами набувають червоного або
фіолетового забарвлення.
Методика. До 0,5 мл 2% розчину лимонної кислоти в оцтовому ангідриді
додають слідову кількість або краплю спиртового розчину аміну і нагрівають
на водяному обігрівачі – з’являється червоне або фіолетове забарвлення.
Позитивну реакцію дають аліфатичні, аліциклічні, мішані, ароматичні і
аліциклічні, а також третинні ароматичні аміни. Солі органічних і неорганічних
кислот з лужними і лужно-земельними металами заважають реакції на відміну
від інших солей. Оскільки реакція дуже чутлива рекомендується паралельно
проводити контрольний дослід. Очевидно через неспецифічність широкого
застосування в фармацевтичному аналізі вона не знайшла, хоча й
рекомендується для підтвердження тотожності ціметідину (2-ціано-1-метил-3-
[2-(5-метилімідазол-4-іл-метилтіо)-етил]-гуанідину).
9. Наявність неподіленої пари електронів надає більшості азотовмісних
органічних сполук не тільки основних властивостей, але й здатність
утворювати комплекси і тому для ідентифікації різноманітних аліфатичних,
аліциклічних, гетероциклічних і мішаних первинних, вторинних, третинних
амінів у фармацевтичному аналізі широко застосовують реакції зі специфічною
групою комплексоутворювачів – загальноалкалоїдними осадовими реактивами.
Загальноалкалоїдні осадові реактиви
1. Реактив Люголя, Вагнера, Бушарда (розчин йоду в калію йодиді).
2. Реактив Драгендофа (розчин бісмуту йодиду в калію йодиді).
3. Реактив Майера (розчин меркурію йодиду в калію йодиді).
4. Реактив Марме (розчин кадмію йодиду в калію йодиді).
5. Реактив Зонненштейна – фосфорномолібденова кислота
(H3PO4
.12MoO3
.2H2O).
6. Реактив Шейблера – фосфорновольфрамова кислота
(H3PO4
.12WoO3
.2H2O).
7. Реактив Бертрана – кремнійвольфрамова кислота (SiO2
.12WoO3
.4H2O).
C2H5OH
KOH
+ +
. HNO3
++
N
O
O
H
HNO3
. HCl
H
CO(CH2)2O N
CH3
CH3
N C4H9
CO(CH2)2O N
CH3
CH3
N C4H9
N
O
O
CH3
CH3
NO CO(CH2)2
N
O
O
C4H9N
O
N
OK
42.
43
8. 5% Розчинтаніну (використовують свіжий розчин).
9. Насичений розчин пікринової кислоти.
1.2.14. Піридиновий цикл
Серед азотовмісних гетероциклічних сполук значну групу складають
похідні піридину і тому піридиновий цикл слід відзначити як специфічну
функціональну групу.
1. Найпростішою реакцією на піридиновий цикл є реакція піролізу. При
нагріванні нікотинової кислоти, її солей, нікотинаміду з безводним карбонатом
натрію відчувається запах піридину (див. карбонові кислоти, 1.2.9.3).
2. Для ідентифікації похідних піридину, у яких вільне α, α'-положення,
проводять реакцію утворення поліметинової основи з 2,4-динітрохлорбензолом.
Методика. До 0,01-0,02 г нікотинової кислоти додають 0,05 г 2,4-
динітрохлорбензолу, 3 мл 95% етилового спирту і кип’ятять протягом 1
хвилини – утворюється сіль піридинію. При додаванні 2 крапель 10% розчину
натрію гідроксиду відбувається розмикання піридинового циклу і утворюється
похідне глутаконового альдегіду - поліметинова основа, що забарвлює розчин в
буро-червоний колір. Далі в результаті гідролізу червоне забарвлення розчину
поступово зникає:
Поліметинові основи утворюються і при використанні таких реагентів як
тіоціанат брому (роданбромідний реактив), тіоціанат хлору, ціанід брому,
хлороформ, хлоралгідрат. В присутності зазначених реагентів в лужному
середовищі відбувається розмикання піридинового циклу і утворюється
глутаконовий альдегід. При наступному додаванні первинних ароматичних
амінів (анілін, новокаїн, сульфацил-натрій) відбувається їх конденсація з
глутаконовим альдегідом і утворюються основи Шиффа забарвлені в жовтий,
оранжевий або червоний колір.
Методика. До 2 мл бромної води додають 2-3 краплі розчину амонію роданіду
(до знебарвлення). В отриманому розчині розчиняють 0,02 г речовини, що
H2O
[OH]
+
CH
HO
C O
H
C O
H
Cl
-
+
+
N
COOH NO2
Cl
NO2
COOH
N
NO2
NO2
NH
COOH
COOH
NO2
NO2
NO2
NO2
NH2
43.
44
досліджується, додають 0,02г новокаїну (або іншого первинного ароматичного
аміну) і краплями 0,1 моль/л розчин натрію гідроксиду до нейтральної реакції –
з’являється жовте забарвлення:
Br2 + NH4SCN → BrSCN + NH4Br
1.2.15. Нітрогрупа
1. Ароматичні нітросполуки – це жовті або білі з жовтуватим відтінком
кристалічні речовини. Якщо в о- або п-положенні до нітрогрупи є група, яка
має рухомий атом гідрогену, то в лужному середовищі внаслідок утворення
ацинітрогрупи з’являється жовте, оранжево-жовте або оранжево-червоне
забарвлення. Наприклад, при розчиненні нітроксоліну в розчині натрію
гідроксиду з’являється оранжево-червоне забарвлення:
Методика. 0,1 г левоміцетину нагрівають з 4-5 мл 10% розчину натрію
гідроксиду – з’являється жовте забарвлення, що переходить при подальшому
нагріванні в оранжево-червоне, відчувається запах амоніаку:
+ NH3
+
Cl2CHCOONa NaOH+
+
NaOH
O2N CH
CHCl2
O
CNH
CH2OHCH
OH
O2N CH
OH
NH2
CH2OHCH
N
ONa
O
C CH CH2OH
OH
OH
+ H2O
+
+ NaOH
N
NO2
OH
N
NNaO O
O
RCH NNHNHR
H2N RRRH2NH2N
COOHCOOHCOOHCOOHCOOHCOOH
N
2
N
SCN
COOH
+
CH
2
COOH
+ BrSCN Br
-
CH
HO
C O
H
NaOH
+ NH2SCN + NaBr
CH
COOH
2
+ 2H2O
основа Шиффа
44.
45
2. Як правило,для виявлення ароматичних нітросполук використовують
реакції відновлення.
Методика. До декількох міліграмів ароматичної нітросполуки додають
2 краплі 10% розчину кальцію хлориду, цинковий пил і нагрівають на водяному
обігрівачі протягом 2 хвилин, додають 2 краплі 5% розчину α-нафтиламіну в
оцтовій кислоті і нагрівають ще 2 хвилини – з’являється червоне забарвлення:
3. При відновленні ароматичних нітросполук цинком в кислому
середовищі утворюються відповідні ароматичні аміни, які можна
ідентифікувати за реакцією утворення азобарвника.
Методика. До декількох крупинок левоміцетину додають 2 мл розведеної
хлороводневої кислоти, 0,1 г цинкового пилу і нагрівають на водяному
обігрівачі протягом 2-3 хвилин. Розчин охолоджують, фільтрують, до фільтрату
додають 3 краплі 0,1 моль/л розчину натрію нітриту і струшують. Отриманий
розчин додають до 3 мл лужного розчину β-нафтолу – з’являється червоне
забарвлення:
1.2.16. Аміди
Аміди – похідні карбонових кислот, у яких гідроксильну групу заміщено
на залишок первинного або вторинного, аліфатичного чи ароматичного аміну:
Окремим випадком амідів є похідні сечовини – уретани, уреїди.
1. Основною якісною реакцією, яка застосовується для ідентифікації
амідів є реакція гідролізу. Гідроліз може бути каталізовано лугами або
R C N
R1
R2
O
R
R
2H
R
O2N ON
NH2
N N
+
NaNO2
HClHCl
Zn
NaOH
Cl
O2N CH CH CH2OH
OH
NH COCHCl2
H2N CH CH2OHCH
OH
NH C CH3
O
CH CH2OHCH
OH
NHCOCH3
NN
OH
ONa
N N
NHCOCH3
OH
CH CH2OHCH
45.
46
кислотами. Якщо амідутворено амоніаком або летким аміном, то при
нагріванні з розчинами лугів виділяється амоніак чи відповідний амін, який
ідентифікують за запахом або посинінням вологого червоного лакмусового
папірця.
Методика. При кип’ятінні 2-3 крапель діетиламіду нікотинової кислоти з 3 мл
розчину натрію гідроксиду виділяється діетиламін, який ідентифікують за
характерним запахом:
Якщо амід утворено леткою органічною кислотою з незначною
молекулярною масою, під час кислотного гідролізу відчувається запах
відповідної кислоти.
Методика. 0,2 г бромізовалу нагрівають з сумішшю 3 мл води і 2 мл
концентрованої сульфатної кислоти – відчувається гострий запах
ізовалеріанової кислоти:
Реакція кислотного гідролізу дозволяє ідентифікувати не лише кислоту,
але й амін.
Методика. 0,1 г парацетамолу обережно кип’ятять з 2 мл розведеної
сульфатної кислоти протягом 2 хвилин – з’являється запах оцтової кислоти:
Після охолодження реакційна суміш дає реакцію на первинні ароматичні
аміни:
Деякі первинні аліфатичні аміни також ідентифікують після кислотного
гідролізу відповідного аміду.
Методика. До 0,1 г бутаміду додають 5 мл розведеної сульфатної кислоти і
кип’ятять протягом 3 хв, потім обережно додають 6 мл 30% розчину натрію
α-бромізовалеріанова кислота
+ (NH4)2SO4 + CO2CH C NH C NH2
Br O O
CH
H3C
H3C
H2SO4
H2O
CH
H3C
H3C
CH
Br
COOH
+ CH3COOH
tº
H2O; H2SO4
HO NH C CH3
O
HO NH2
N N
HCl
NaNO2
+
Cl
-
NaOH
NH2
OH OH
OH ONa
N N
ONa
COONaC
O
N(C2H5)2
N
N
NaOH
+ HN (C2H5)2
46.
47
гідроксиду – наповерхні утворюються масні краплі бутиламіну, який має
характерний запах:
2. Відомі реакції, які дозволяють розрізнити незаміщені аміди
ароматичних і аліфатичних кислот. Більшість ароматичних амідів на відміну від
аліфатичних при взаємодії з перекисом водню утворюють гідроксамові
кислоти. Більшість аліфатичних амідів перетворюються на гідроксамові
кислоти при обробці гідроксиламіном в водному або спиртовому розчині, в той
час як ароматичні аміди в цих умовах реагують набагато важче:
Ароматичні аміди. До суспензії 50 мг аміду в 2-3 мл води при
перемішуванні додають 4-5 крапель 6% розчину перекису водню і нагрівають
до кипіння. Якщо при цьому речовина повністю не перейде у розчин, додають
ще декілька крапель розчину перекису водню. Після охолодження додають
краплю 4% розчину феруму (III) хлориду. Якщо протягом 1 хв не з’явиться
виразне фіолетове забарвлення, то розчин злегка підігрівають, не доводячи до
кипіння. Помітне на холоду, слабке червонувате забарвлення переходить в
інтенсивне синьо-червоне, при подальшому нагріванні – коричневе; поступово
випадає лапатий темно-коричневий осад.
Аліфатичні аміди. До 50 мг аміду додають 1 мл насиченого при
кімнатній температурі розчину гідрохлориду гідроксиламіну в спирті і суміш
кип’ятять протягом 3 хв. Після охолодження додають 1-2 краплі 5% розчину
феруму (III) хлориду – виникає характерне для гідроксамових кислот червоне
або червоно-фіолетове забарвлення.
3. Амідам, утвореним первинними амінами або амоніаком, притаманне
явище амідо-імідольної таутомерії, за рахунок якої вони мають слабкі кислотні
властивості й здатні утворювати комплекси з солями важких металів,
наприклад, купруму:
Методика. 0,05 г кальцію пангамату розчиняють в 5 мл розчину натрію
гідроксиду і фільтрують. До фільтрату додають 3 краплі розчину купруму
сульфату – з’являється синє забарвлення.
ArCONH2 + H2O2 ArCONHOH + H2O
AlcCONH2 + H2NOH . HCl AlcCONHOH + NH4Cl
R' N C R
R C N R'
O
Cu
OCuSO4
R C N R'
ONa
NaOH
R C N
OH
R'R C NHR'
O
C4H9NH2
. H2SO4 + 2NaOH C4H9NH2 + Na2SO4 + 2H2O
+ CO2 + C4H9NH2
. H2SO4
H2SO4
H2O
CH3 SO2 NH C NH C4H9
O
CH3 SO2 NH2
47.
48
Для підтвердження тотожностіамідів рекомендується також проводити
реакцію з солями кобальту.
Методика. Розчиняють 0,1 г основи лідокаїну (2-діетиламіноацето-2`,6`-
ксилідину) в 1 мл 96% етанолу і додають 0,5 мл 10% розчину кобальту (II)
нітрату – утворюється синьо-зелений осад.
4. Такий амід як сечовина, а також її ацильні похідні – уреїди дають
біуретову реакцію.
Методика. Нагрівають 0,5 г сечовини в пробірці до розплавлення, прогрівають
поки рідина стане мутною і охолоджують. Розчиняють плав у суміші 10 мл
води і 1 мл 2 моль/л розчину натрію гідроксиду і додають 0,05 мл розчину
купруму сульфату – утворюється червоно-фіолетове забарвлення.
Ця реакція є спільною для сполук, що містять не менш двох груп:
При нагріванні бромізовалу з розчином CuSO4 в лужному середовищі
з'являється рожево-червоне або (при надлишку CuSO4) червоно-фіолетове
забарвлення:
5. До похідних сечовини можна віднести сполуки, що містять гуанідинове
угрупування:
Специфічною реакцією на гуанідини є реакція Сакагучі.
Методика. До 5 мл 0,5% розчину стрептоміцину сульфату додають 1 мл 0,5%
розчину α-нафтолу в 40% спирті. Суміш охолоджують до 15о
С і додають
3 краплі 5% розчину гіпоброміду натрію – з’являється фіолетово-червоне
забарвлення.
C NH2
O
або, C
S
NH2 C NH2
NH
C NH2
NH
NHR
tº
CH C NH C NH2
Br O O
R NaOH R CH C ONa
OH O
+ C O
H2N
H2N
NH3 + NH C O
NaOH
H2N C NH2
O
ізоціанова
кислота
O O
OH
H
O
C NH2
Cu
NHC
N
H2N C
NH
CN
CuSO4; NaOH tº
H2N C NH C NH2
O O
H2N C NH2
O
+HN C O
,
48.
49
1.2.17. Іміди
До імідіввідносять сполуки загальної формули:
На практиці частіше доводиться зустрічатися з циклічними імідами:
1. Іміди гідролізуються важче ніж аміди. Для їх гідролізу потрібні більш
жорсткі умови – тривале кип’ятіння з 30% розчином натрію гідроксиду або
сплавлення з кристалічним натрію гідроксидом або натрію карбонатом. Ця
реакція характерна, наприклад, для барбітуратів. При сплавленні з їдкими
лугами барбітурати розкладаються з утворенням солей діалкіл- або
арилалкілпохідних оцтової кислоти, натрію карбонату й амоніаку, який
ідентифікують за запахом або посинінням вологого червоного лакмусового
папірця:
N
N
O
O
R
R1
O
H
H
+ 5NaOH CH
R
R1
COONa + 2NH3 + 2Na2CO3
При наступному підкисленні виділяються бульбочки газу (CO2) і вільні
похідні оцтової кислоти, які мають специфічний запах:
Na2CO3+2HCl H2O+CO2 +2NaCl
CH
R
R1
COONa + HCl CH
R
R1
COOH + NaCl
2. Наявність імідної групи за рахунок імідо-імідольної таутомерії надає
сполуці слабких кислотних властивостей:
Як і більшість органічних сполук, що мають кислотні властивості іміди
ідентифікують за реакціями утворення комплексних солей з іонами важких
металів. Найчастіше використовують реакцію з солями Co2+
.
C
O
NH
C
O
R
R C NH C R'
O O
C
C
O
R N
ONa
NaOH
C
C
O
R N
OH
C
O
NH
C
O
R + H2O
49.
50
Методика. а) 0,05г кислотної форми барбітурату розчиняють у 2 мл 95%
спирту, додають 1 краплю розчину кальцію хлориду, 2 краплі розчину кобальту
нітрату, 2 краплі розчину натрію гідроксиду – з’являється синьо-фіолетове
забарвлення:
б) реакція Цвіккера. Речовину, що досліджується розчиняють в 1 мл
суміші піперидину і хлороформу (1:9) і додають 0,5 мл розчину купруму
сульфату (розчин Фелінга №1, розбавлений у 10 разів). При енергійному
струшуванні хлороформний шар набуває забарвлення: в присутності
барбітурових кислот – фіолетового, тіобарбітурових кислот і тіоурацилів –
зеленого, гідантоїнів – синього. Слід мати на увазі, що реакцію Цвіккера дають
також пурини, деякі сульфаміди, сахарин і ноксирон;
в) до 0,1 г теоброміну додають 2 мл 0,1 моль/л розчину натрію
гідроксиду, струшують протягом 2-3 хвилин і фільтрують. До фільтрату
додають 3 краплі 2% розчину кобальту хлориду і перемішують – з’являється і
швидко зникає інтенсивне фіолетове забарвлення і зразу ж утворюється осад
сірувато-блакитного кольору:
Розрізняють барбітурати за допомогою реакції з купруму (II) сульфатом в
присутності гідрокарбонату і карбонату калію (табл. 1.1).
Методика. 0,1 г речовини збовтують протягом 1-2 хвилин з 1 мл 1% розчину
натрію гідроксиду (кислотні форми барбітуратів) або розчиняють у 1 мл води
(сольові форми барбітуратів), додають 2 краплі розчину купруму сульфату і 4
краплі розчину суміші карбонату і гідрокарбонату калію.
NaCl2+Co
+2
2
O
O
N
N N
N CH3
CH3
CoCl2
2
O
O
N
N N
N CH3
CH3
Na
+
H2O+
Na
CH3
CH3
N
NN
N
O
O
NaOH+
CH3
CH3
N
NN
N
O
O
H
HN
HN
O
O
O
R
R1
CoCl2
CaCl2; NaOH
NH
NR
R1
O
O
O Co
O
O
R
R1HN
O
O
R
R1
O
N
50.
51
Таблиця 1.1
Речовина Результатреакції
1 2
Барбаміл Осад рожево-бузкового кольору, що не змінюється при стоянні
Барбітал, барбітал-натрій Синє забарвлення, потім осад червоно-бузкового кольору
Бензонал Сіро-блакитне забарвлення, що не змінюється при стоянні
Гексенал Блакитне забарвлення, що переходить в синє, при стоянні
випадає білий осад
Тіопентал-натрій Жовто-зелене забарвлення з зависом осаду, що не змінюється
при стоянні
Фенобарбітал Осад блідо-бузкового кольору, що не змінюється при стоянні
Етамінал-натрій Осад блакитного кольору, що не змінюється при стоянні
Окрім солей купруму й кобальту іміди дають комплекси й з солями інших
важких металів, наприклад, меркурію. Досить часто для ідентифікації сполук,
що містять імідну групу застосовують реакції з солями аргентуму.
Методика. а) 0,05 г теоброміну розчиняють у суміші 3 мл води і 6 мл розчину
натрію гідроксиду, додають 1 мл розчину амоніаку і 2 мл 5% розчину
аргентуму нітрату – після струшування утворюється густа желатиноподібна
маса, яка розріджується при нагріванні до 80оС і знову застигає при
охолодженні;
б) при додаванні до крупинки або декількох крапель розчину
рибофлавіну 3-4 крапель розчину аргентуму нітрату утворюється комплексна
сполука оранжево-червоного кольору.
Здатність утворювати комплекси з солями важких металів
використовується для кількісного визначення імідів методами аргентометрії
(барбітурати) і непрямої алкаліметрії (теобромін, рибофлавін).
1.2.18. Гідразини, гідразиди, гідразони
Гідразини – сполуки, в яких один з атомів гідрогену молекули гідразину
заміщено на алкільний або арильний радикал: R–NH–NH2.
До гідразидів відносять сполуки, в яких один з атомів гідрогену в
молекулі гідразину заміщено на залишок карбонової кислоти:
Гідразони – продукти конденсації гідразинів або гідразидів з альдегідами
або кетонами, в яких два атоми гідрогену при одному з атомів азоту заміщено
на залишок альдегіду або кетону:
1. Залишок гідразину надає гідразинам і гідразидам відновних
властивостей.
R C
O
NH NH R' , де R'=H; Alc; Ar
, де R'=H; Alc; Ar; AcylR C N NH R'
H
51.
52
Методика. Розчиняють 0,1г фенелзину (фенетилгідразину гідросульфату) в
5 мл води, підлуговують 5 моль/л розчином натрію гідроксиду і додають 1 мл
реактиву Фелінга – утворюється цеглинно-червоний осад.
2. Гідразидну групу можна розглядати як окремий випадок амідів. За
рахунок утворення імідольної форми гідразиди дають комплексні солі з іонами
важких металів – аргентуму або купруму (II). При наступному нагріванні
відбувається окиснення гідразину і відновлення іонів аргентуму до металічного
срібла , а іонів купруму (II) до оксиду купруму (I).
Методика. 0,1 г ізоніазиду розчиняють в 5 мл води і додають 4-5 крапель
розчину купруму сульфату – виділяється блакитний осад, при струшуванні
розчин також стає блакитним. При нагріванні розчин і осад стають світло-
зеленого, потім жовто-зеленого кольору, виділяються бульбочки газу:
3. Якщо альдегіди ідентифікують по реакції утворення гідразонів з
гідразином, то так само гідразини і гідразиди ідентифікують по реакції з
альдегідами.
Методика. а) Розчиняють 0,1 г ізоніазиду в 2 мл води, додають теплий розчин
0,1 г ваніліну в 10 мл води, через деякий час, після потирання стінок пробірки
скляною паличкою – утворюється жовтий осад. Його відфільтровують,
перекристалізовують в 35 мл 70% спирту, висушують при 100-105о
С і
визначають температуру плавлення (226-231о
С):
б) 0,1 г ізоніазиду розчиняють в 2 мл води, додають 1 мл розчину натрію
гідроксиду, кип’ятять протягом 1 хв і охолоджують. До отриманого розчину
додають 1 мл розчину п-диметиламінобензальдегіду і 3 мл розведеної
хлороводневої кислоти – виникає жовто-оранжеве забарвлення:
N
CONHNH2
N
C
OH
N NH2
CuSO4
NN
C
O Cu O
C
NNH2 H2NN
H2O
tº
N
COOH
+N2 +Cu2O
N
CONHNH2
+
OH
OCH3
C
H
O
N
CONHN CH
OCH3
OH
+ H2O
N(CH3)2C
O
H
C
O
ONa
C
O
NH NH2
NN
2
NH2
NH2
+
NaOH
52.
53
4. Для ідентифікаціїгідразонів застосовують реакцію гідролізу, під час
якого утворюється відповідний альдегід, котрий можна ідентифікувати за
характерним запахом.
Методика. 0,05 г фтивазиду нагрівають з 10 мл розведеної хлороводневої
кислоти – з’являється сильний запах ваніліну:
1.2.19. Тіоли, тіони, тіоефіри, тіоаміди
До цієї групи відносять сполуки, які містять функціональні групи:
1. Найбільш загальною реакцією, яка використовується для ідентифікації
сірковмісних сполук є реакція окисної мінералізації з наступною
ідентифікацією утвореного сульфат-іона взаємодією з барію хлоридом.
Методика. а) До 0,2 г етоксиду (N,N`-[ди-(п-етоксифеніл)]тіосечовини)
додають 5 мл розведеної нітратної кислоти і доводять до кипіння, потім
охолоджують, фільтрують через беззольний фільтр. Фільтрат дає характерну
реакцію на сульфати;
б) до 20 мг пропілтіоурацилу додають 8 мл бромної води і перемішують
протягом 5 хв. Розчин кип’ятять до знебарвлення, дають охолонути і
фільтрують. Додають 2 мл розчину барію хлориду – виникає білий осад.
2.Для ідентифікації сірковмісних сполук зі ступенем окиснення сірки (-2),
застосовують реакції кислотного або лужного гідролізу. При цьому
утворюється сірководень або сульфіди, що їх підкисленням переводять у
сірководень, який ідентифікують за запахом або реакцією з плюмбуму
ацетатом.
Методика. а) 0,1 г тіофосфаміду вміщують у пробірку, додають 3 мл
розведеної хлороводневої кислоти і кип’ятять у полум’ї пальника –
відчувається запах сірководню;
;;; R C NHR'
S
R S R'R C R'
S
R SH
+ NH2NH2
. 2HCl+
N
COOH
H2O
N
CONHN CH
OCH3
OH
OH
OCH3
C
H
O
HCl, tº
N(CH3)2
N(CH3)2
N(CH3)2
N(CH3)2CHN
N CH
Cl
-
+
HCl
CHN
NH CH
53.
54
б) кип’ятять 0,1г моносульфіраму (тетраетилтіоурацилу моносульфід) з 2
моль/л розчином хлороводневої кислоти – пари, що виділяються, забарвлюють
фільтрувальний папір, змочений розчином плюмбуму ацетату, в чорний колір;
в) 0,05 г метіоніну нагрівають у пробірці з 5-6 краплями 30% розчину
натрію гідроксиду до отримання сплаву. До охолодженого сплаву додають 5 мл
води і підкислюють розведеною сульфатною кислотою – з’являється запах
сірководню і меркаптану:
г) 0,2 г тіопентал-натрію розчиняють в 5 мл розчину натрію гідроксиду,
додають 2 мл розчину ацетату плюмбуму і кип’ятять – випадає темний осад.
Після охолодження і підкислення концентрованою хлороводневою кислотою
виділяється сірководень, який ідентифікують за запахом і потемнінням
фільтрувального паперу, змоченого розчином ацетату плюмбуму:
3. Одним з реактивів, що широко застосовується для ідентифікації тіолів
є нітропрусид натрію [Na2Fe(NO)(CN)5].
Методика. а) 0,05 г меркаптопурину вміщують у пробірку, розчиняють у
0,5 мл розчину натрію гідроксиду, розбавляють водою до 2 мл і додають 0,5 мл
свіжоприготованого розчину натрію нітропрусиду; пробірку струшують –
з’являється жовто-зелене забарвлення, що переходить при підкисленні
розведеною хлороводневою кислотою в темно-зелене;
б) 0,02 г мерказолілу розчиняють у 1 мл води, додають 1 мл розчину
натрію гідроксиду і збовтують. До отриманого розчину додають 3 краплі
розчину натрію нітропрусиду – через декілька хвилин з’являється жовте
+ 2NH3 + 2Na2CO3 + 2CH3COONa
HC
CH3
CH2
CH
CH2 CH3
COONa
H5C2
+PbS
N
N
NaS
H O
O
C2H5
CH
CH3
CH2 CH2 CH3 + 6NaOH + (CH3COO)2Pb
чорний
+ CH3SNa + Na2S + CH3OH + 2NH3
+CH2 CH2 CH C
ONH2
2HO
ONa
Na2S + CH3SNa + 2H2SO4 H2S + CH3SH + Na2SO4 + NaHSO4
+ 5NaOH2H3C S CH2 CH2 CH C
O
OH
NH2
H2S + PbCl2PbS + 2HCl
54.
55
забарвлення, що переходитьу зелене. Після додавання 1 мл оцтової кислоти
забарвлення переходить у світло-синє.
Іноді для виділення сірководню речовину відновлюють натрію
форміатом.
Методика. Змішують у пробірці 10 мг тіогуанідину з 10 мг форміату натрію і
обережно нагрівають до розплавлення – газ, що виділяється, забарвлює папір,
змочений плюмбуму ацетатом, в темний колір.
4. Тіоли, в порівнянні зі спиртами, мають підвищену кислотність і дають
комплексні солі з іонами важких металів. Таку саму реакцію дають також
тіоаміди і деякі тіони. В утворенні комплексів беруть участь неподілені
електронні пари сульфуру.
Методика. а) 0,01 г мерказолілу розчиняють в 1 мл води, додають 2 краплі
розчину аргентуму нітрату – утворюється білий осад, нерозчинний у воді й
надлишку нітратної кислоти:
Ця ж реакція лежить в основі кількісного визначення мерказолілу
методом непрямої алкаліметрії.
б) розчиняють 10 мг етіонаміду (2-етилпіридин-4-карботіоамід) в 5 мл
метанолу і додають 5 мл 0,1 моль/л розчину аргентуму нітрату – випадає темно-
коричневий осад;
в) розчиняють 20 мг меркаптопурину в 20 мл 96% етанолу, нагрівають до
60о
С і додають 1 мл розчину меркурію (II) ацетату в 96% етанолі – утворюється
білий осад.
Досить часто для ідентифікації тіосполук застосовують реакцію з солями
купруму (II).
г) розчиняють 0,1 мл димеркаптолу ((R,S)-2,3-димеркаптопропанол-1) в 5
мл води і додають 2 мл розчину сульфату купруму (II) – утворюється синьо-
чорний осад, який швидко переходить у темно-сірий;
д) розчиняють 0,1 г моносульфіраму в суміші 0,15 мл розчину купруму
(II) сульфату і 5 мл 96% етанолу й випарюють на водяному обігрівачі досуха;
залишок розчиняють у хлороформі – з’являється жовто-коричневе забарвлення.
5. Неподілені електронні пари сульфуру надають сполуці здатність
утворювати комплекси, зокрема з тетрайодбісмутатом калію (реактивом
Драгендорфа).
Методика. До 5 мг карбімазолу (етил-3-метил-2-тіоксо-4-імідазоліно-1-
карбоксилат) додають 0,05 мл розведеного розчину калію йодбісмутату –
утворюється яскраво-червоне забарвлення.
+ HNO3+ AgNO3
N
SH
CH3
N
N
SAg
CH3
N
55.
56
1.2.20. Сульфокислоти, сульфаміди
Сульфокислоти– органічні сполуки загальної формули R–SO3H.
Сульфокислоти належать до числа найбільш сильних органічних кислот.
Сульфаміди – похідні сульфокислот, в яких гідроксильну групу заміщено на
залишок аміну або аміду. Вони мають загальну формулу R–SO2NH–R`.
1. Як і інші сірковмісні органічні сполуки сульфокислоти й сульфаміди
ідентифікують по реакції на сульфат-іон, після мінералізації з сумішшю для
спікання.
2. Оскільки обидва класи сполук проявляють кислотні властивості,
ідентифікують їх також по реакції з солями важких металів.
Методика. До 10 мл розчину хініофону (1:1000) додають 5 мл розчину барію
хлориду – випадає жовтий осад, розчинний у 2 мл розведеної хлороводневої
кислоти.
Найбільш численну групу похідних сульфамідів, які застосовуються в
хіміотерапії, становлять похідні п-амінобензолсульфаміду (сульфаніламіду),
утворенні шляхом заміщення в ньому атомів гідрогену аміно- або сульфамідної
групи:
Їх хімічні властивості визначаються, головним чином, реакціями цих груп.
Наявність первинної ароматичної аміногрупи зумовлює реакції утворення
азобарвника, основ Шиффа з альдегідами, галогенопохідних (див. аміни,
1.2.13).
Наявність сульфамідної групи зумовлює реакції з солями важких металів,
з яких найчастіше застосовують феруму (III) хлорид; кобальту (II) хлорид і
купруму (II) сульфат (табл. 1.2).
Методика. 0,1 г речовини збовтують з 3 мл 0,1 моль/л розчину натрію
гідроксиду і фільтрують. Фільтрат розливають у три пробірки і додають по 2-3
краплі розчинів феруму (III) хлориду; кобальту (II) хлориду; купруму (II)
сульфату:
RNH SO2NHR'
+ Na2SO42
CuSO4
H2N
H2N SO2NR
Na
H2N SO2 N R
Cu
RNSO2
56.
57
Таблиця 1.2
Кольорові реакціїдеяких лікарських речовин, які містять
сульфамідну групу з солями важких металів
Колір осаду або розчину
Речовина хлорид феруму (III) хлорид кобальту(II) сульфат купруму(II)
Стрептоцид Жовтий розчин Блакитний з синюва-
тим відтінком роз-
чин
Зеленуватий з бла-
китним відтінком
розчин
Стрептоцид
розчинний
Червонуватий
розчин
Рожевий розчин Блакитний з зелену-
ватим відтінком осад
Сульфацил-натрій
Червонуватий
розчин
Рожевий розчин
Блакитно-зеленува-
тий осад, який не
змінюється при
стоянні
Сульгін Жовтий розчин Рожевий розчин Блакитний розчин
Уросульфан Жовтий розчин Рожевий розчин Світло-зелений роз-
чин
Норсульфазол Світло-оранжевий
осад
Бузковий осад,який
швидко переходить в
синьо-фіолетовий
Брудно-фіолетовий
осад, що переходить в
темно-ліловий
Етазол Світло-оранжевий
розчин
Рожевий розчин,
білий осад
Трав’янисто-зелений
осад, що переходить в
чорний
Сульфадимезин Світло-оранжевий
розчин
Рожевувато-бузко-
вий осад
Жовтувато-зелений,
осад, що швидко
переходить в корич-
невий
Сульфален Жовтий розчин Рожевий розчин Брудно-зелений осад,
що поступово перехо-
дить в зеленувато-
блакитний
Сульфамонометоксин Жовтий розчин Рожево-малиновий
розчин
Сірувато-зелений
осад, не змінний
протягом 30 хвилин
Сульфадиметоксин Жовтий розчин Яскраво-рожевий з
лиловим відтінком
осад
Зелений осад
Сульфазин Жовтий розчин Бузковий осад Брудно-зелений з
жовтуватим відтін-
ком осад; через 5-7
хвилин забарвлення
переходить у бруд-
но-бузкове
Сульфапіридазин Жовтий розчин Рожевий розчин Трав’янисто-зелений
осад
Фталазол Жовтий розчин Рожевий розчин Брудно-зеленувато-
сірий осад
Фтазин Жовтий розчин Рожевий розчин Зеленувато-блакит-
ний осад
57.
58
Кислотні властивості сульфамідноїгрупи використовуються для
кількісного визначення сполук цього ряду методоми аргентометрії та
алкаліметрії в ацетоновому середовищі з індикатором тимолфталеїн.
1.2.21. Фосфорорганічні сполуки
(органічні фосфоровмісні сполуки)
Органічні сполуки фосфору ідентифікують, як правило, за реакцією на
фосфат-іон після гідролізу або мінералізації.
Наприклад, фосфотіамін, розчинений в розведеній нітратній кислоті дає
характерну реакцію на фосфорну кислоту з розчином амонію молібдату.
Кокарбоксилаза дає позитивну реакцію на фосфорну кислоту з розчином
амонію молібдату після 5 хв кип’ятіння з концентрованою нітратною кислотою,
в результаті якого відбувається гідроліз речовини (див. фосфати, 1.1.2.12.3).
Методика. 0,1 г тіофосфаміду вміщують в колбу К’єльдаля ємністю 50 мл,
додають 2 мл концентрованої сульфатної кислоти, 1 мл концентрованої
нітратної кислоти і кип’ятять до знебарвлення розчину й видалення окислів
азоту. Після охолодження додають 3 мл води і обережно нейтралізують
концентрованим розчином амоніаку за лакмусом; 2 мл отриманого розчину
підкислені 1 мл розведеної нітратної кислоти, дають характерну реакцію на
фосфати з розчином амонію молібдату.
58.
59
2. Визначення чистотилікарських речовин
Практично всі лікарські речовини мають у своєму складі сторонні домішки.
Їх наявність може бути зумовлена такими причинами:
– недостатньою чистотою вихідних продуктів, які застосовуються при синтезі
або вилученні з сировини;
– утворенням побічних продуктів в процесі синтезу;
– екстракцією сторонніх речовин з рослинної сировини;
– екстракцією катіонів металів з матеріалу реакторів або намолом робочого
органу млинів;
– розкладом лікарської речовини при неправильному зберіганні або
забрудненням продуктами розкладу таропакувальних матеріалів.
В залежності від характеру та властивостей домішки можуть бути поділені на
дві групи:
– домішки, які характеризують ступінь очистки препарату;
– домішки, які впливають на фармакологічну дію.
В кожній окремій фармакопейній статті (далі – монографії) наводиться
перелік показників, за якими встановлюється чистота лікарської речовини
(зовнішній вигляд, розчинність, температура топлення або кипіння,
забарвленість та мутність розчину та ін.).
Невідповідність лікарської речовини навіть одному з показників,
закладених у нормативно-технічній документації, свідчать про її
недоброякісність.
2.1. Визначення забарвленості та мутності
Лікарські речовини можуть бути забруднені домішками, поява яких
зумовлена зміною їх фізичних та хімічних властивостей під впливом вологості,
світла, кисню, вуглекислого газу та інших факторів навколишнього
середовища.
Наявність домішок частіше за все викликає зміну забарвлення розчинів
лікарських речовин або розчинності у воді та інших розчинниках. Контроль за
прозорістю розчинів лікарських речовин регламентується вказівками загальної
фармакопейної статті та монографій. Ступінь мутності визначають порівнянням
рідини або розчину, що досліджується, з розчинником або еталонами. Як
еталон використовують, наприклад, суспензії, що утворюються в результаті
реакції водних розчинів гідразину сульфату та гексаметилентетраміну. Таких
еталонів, з якими порівнюють ступінь мутності, може бути, наприклад, 4.
Розчин вважається прозорим, якщо при його розгляданні неозброєним оком не
спостерігається наявність нерозчинних часток. Порівняння проводять з
розчинником. Спостереження здійснюють у світлі, що проходить через розчин,
на темному фоні.
59.
60
Для визначення ступенюмутності забарвлених рідин частину рідини, що
досліджується, фільтрують і вміщують до компаратору, а поруч ставлять
пробірку з нефільтрованою рідиною. За нею ставлять пробірку з розчинником,
а за пробіркою з фільтрованою рідиною по черзі вміщують пробірки з
еталонами. Вибирають той, який має однаковий ступінь мутності з
нефільтрованою рідиною.
У разі визначення припустимої межі забарвлених домішок порівняння
проводять з еталонами забарвленості. Приготування еталонів наведено у
відповідній загальній фармакопейній статті.
Для приготування вихідних розчинів використовуються кобальту (II)
хлорид, калію дихромат, купруму (II) сульфат, феруму (III) хлорид, які
розчиняють в 0,1 моль/л розчині сульфатної кислоти.
Змішуванням вихідних розчинів у певних співвідношеннях готують
основні розчини. З основних розчинів при необхідності готують еталони, з
якими порівнюють забарвлення досліджуваної рідини. Таблиці приготування
еталонних розчинів наводяться у відповідній фармакопейній статті або додатку.
Порівняння забарвленості рідин здійснюють у відбитому світлі на білому фоні.
Безбарвною вважається рідина, яка за забарвленням не відрізняється від води і
розчин речовини, який за забарвленням не відрізняється від відповідного
розчинника.
При визначенні ступеню забарвленості та мутності розчинів для
порівняння беруть однакові об’єми еталону та рідини, що досліджується (5-
10 мл). Пробірки, в яких проводиться визначення, повинні бути одного
діаметру та виготовлені з однакового скла.
2.2. Визначення кислотності, лужності та pH середовища
Невідповідність лікарської речовини за цими показниками може бути
наслідком присутності домішок більш кислотного або основного характеру, ніж
сама речовина.
Подібні домішки можуть утворюватися при отриманні або зберіганні
лікарських засобів (домішка мінеральних кислот, продуктів гідролізу, вплив
лужності скла, поглинання речовиною вуглекислого газу з повітря і т. ін.).
Контроль кислотності, лужності, pH середовища регламентується
відповідним розділом монографій та може здійснюватися різними способами:
- за зміною забарвлення кислотно-основних індикаторів (pH-індикаторів);
- кислотно-основним титруванням;
- визначенням величини pH, за методом, наведеним в загальній
фармакопейній статті “Визначення pH”. Таке визначення є обов’язковим для
всіх ін’єкційних розчинів.
60.
61
2.3. Визначення домішокіонів
В фармакопейному аналізі існують два методи визначення вмісту домішок:
1. Порівняння зі стандартом. При цьому застосовується порівняння
інтенсивності забарвлення або помутніння, які утворюються у розчині, що
досліджується, та еталонному (стандартному) розчині при однакових
умовах.
2. Визначення межі відсутності забарвлення або помутніння. При цьому про
наявність або відсутність домішок судять по відповідному позитивному
або негативному результату хімічної реакції на дану домішку в розчині
певної концентрації.
Для визначення домішок використовують реакції з високою чутливістю
та специфічністю.
Чутливість реакції виражається мінімальною кількістю речовини (іона),
яка може бути визначена з допомогою цього реагенту при певних умовах
проведення реакції.
Специфічними є реакції, які дозволяють знаходити малі кількості
речовини в присутності інших сполук. Специфічність здебільшого залежить від
вибору реагенту та оптимальних умов проведення реакції.
Для визначення допустимої межі домішок застосовують еталонні
розчини, які містять точно встановлену концентрацію іона, що визначається.
Розчини для дослідження готують за методиками зазначеними у
відповідних монографіях. Вміст лікарської речовини в цих розчинах
розраховують так, щоб при наявності в них гранично можливої кількості
домішки її масова доля в досліджуваному та еталонному розчинах були
однакові. Завдяки цьому якісні дослідження мають кількісне значення.
У практиці фармакопейного аналізу іноді доводиться визначати
допустиму межу вмісту домішки без зазначення концентрації розчину, що
досліджується. В цьому випадку для розрахунків використовують формулу:
A
b
C
⋅
=
100
, (2.1)
де С – концентрація розчину речовини, що аналізується, %;
b – концентрація еталонного розчину, %;
А – допустимий фармакопейною статтею вміст домішки, %.
Допустиму межу вмісту домішки в 1 мл розчину субстанції, що
досліджується, виготовленому за методикою відповідної монографії,
розраховують за формулою:
V
mC
X
⋅
⋅
=
100
, (2.2)
де С – гранично допустимий (за ФС) вміст домішки в лікарській
речовині, %;
m – маса наважки лікарської речовини, г;
61.
62
V – об’ємрозчинника, використаного для розчинення лікарської
речовини, мл.
Методики для визначення домішок іонів, які найчастіше зустрічаються в
лікарських речовинах (Cl-, SO4
2-, Ca2+
, Fe3+
та ін.), як правило, наводять у
загальній фармакопейній статті “Випробування на чистоту та допустимі межі
вмісту домішок”. У цій же статті наводять методику приготування еталонних
розчинів та їх концентрацію.
При визначенні чистоти лікарських речовин треба суворо дотримуватися
таких загальних зауважень:
1. Вода та всі реактиви повинні бути вільними від іонів, на вміст яких
проводять дослідження.
2. Пробірки, в яких проводять дослідження, повинні бути безбарвними та
однакового діаметру.
3. Наважки для виготовлення еталонних розчинів відважують з точністю до
0,001 г.
4. Еталонні розчини (друге або третє розведення, наприклад Б і В)
виготовляють перед застосуванням.
5. Спостереження муті та опалесценції розчинів проводять у світлі, що
проходить через розчин на темному фоні, а забарвлення – при денному
відбитому світлі на матово-білому фоні.
6. Додавання реактивів до досліджуваного та еталонного розчинів повинно
проводитися одночасно і в однакових кількостях.
7. У випадку, коли у відповідній фармакопейній статті вказано, що у цій
концентрації розчину не повинна визначатися та чи інша домішка, діють
таким чином. До 10 мл розчину, що досліджується, додають усі реактиви,
крім основного. Потім розчин розливають у дві пробірки. До однієї з них
додають основний реактив і обидва розчини порівнюють між собою. Між
ними не повинно бути помітної різниці.
2.3.1. Дослідження на хлориди
Розчини хлоридів, у залежності від їх концентрації, утворюють з
розчином аргентуму нітрату в нітратнокислому середовищі білий сирнистий
осад, білу муть або опалесценцію:
Гранична чутливість реакції – 0,0001 мг хлор-іона в 1 мл розчину.
Підкислення іншими кислотами не дозволяється, оскільки в цьому
випадку можуть утворитися нерозчинні солі аргентуму.
Методика. До 10 мл розчину лікарського засобу, приготовленого за вимогами
монографії, додають 0,5 мл нітратної кислоти, 0,5 мл розчину аргентуму
нітрату, перемішують і через 5 хвилин порівнюють з еталонним розчином
(10 мл), в який додано таку ж кількість реактивів, як і в розчин, що
досліджується.
Cl
-
+ Ag+ AgCl
62.
63
2.3.2. Дослідження насульфати
Розчини сульфатів, у залежності від їх концентрації, утворюють з
розчинами солей барію (BaCl2, Ba(NO3)2) білий осад або муть, які не зникають
від додавання розведеної хлороводневої кислоти:
Реакцію проводять у середовищі розведеної хлороводневої кислоти, щоб
запобігти утворенню осадів BaCO3 або BaHPO4.
Гранична чутливість реакції – 0,003 мг сульфат-іона в 1 мл розчину.
Методика. До 10 мл розчину лікарського засобу, приготовленого за вимогами
монографії, додають 0,5 мл розведеної хлороводневої кислоти, 1 мл розчину
барію хлориду, перемішують і через 10 хвилин порівнюють з еталоном (10 мл),
до якого додано таку ж кількість реактивів, як і в розчин, що досліджується.
2.3.3. Дослідження на солі амонію
У залежності від властивостей лікарської речовини та допустимої
концентрації домішки солей амонію її визначення проводиться одним з двох
методів.
Метод I ґрунтується на здатності солей амонію утворювати з реактивом
Несслера (лужний розчин калію тетрайодмеркурату (II)) жовто-бурий осад або
жовте забарвлення у залежності від концентрації:
Гранична чутливість реакції – 0,0003 мг іона амонію в 1 мг розчину.
Методика. До 10 мл розчину лікарського засобу, приготовленого за вимогами
монографії, додають 0,15 мл реактиву Несслера, перемішують і через 5 хвилин
порівнюють з еталонним розчином (10 мл), що містить таку ж кількість
реактивів, як і розчин, що досліджується.
У лікарських речовинах, які містять лужноземельні та важкі метали,
визначення проводять після розчинення в мінімальній кількості води та
додавання 2 мл розчину натрію гідроксиду та 2 мл розчину натрію карбонату.
Розчин розбавляють водою до необхідної концентрації, збовтують і
фільтрують. Визначення домішки проводять в 10 мл фільтрату за основною
методикою.
В лікарських речовинах, які містять більше 0,03% домішки феруму,
визначення проводять після його зв’язування у безбарвний комплекс з натрію-
калію тартратом:
У цьому випадку визначення проводять після додавання до 10 мл розчину
лікарського засобу 2 крапель розчину натрію гідроксиду та 3 мл 20% розчину
SO4
2- + Ba2+ BaSO4
[FeC4H2O6]+ + 2H+Fe3+ + C4H4O6
2-
+
NH2
Hg
Hg
I
I
NH4
+
+ 2[HgI4]2- + 2OH
- I
-
+ 5I
-
+ 2H2O
63.
64
натрію-калію тартрату. Одержанийрозчин перемішують, додають 0,15 мл
реактиву Несслера та порівнюють з еталонним розчином.
Метод II ґрунтується на здатності солей амонію під дією натрію
гідроксиду виділяти амоніак, який визначають за запахом або посинінням
вологого червоного лакмусового папірця:
Гранична чутливість реакції – 0,003 мг іона амонію в 1 мл розчину.
Методика. 5 мл розчину лікарського засобу, приготовленого за вимогами
монографії, вносять в конічну колбу ємністю 25 мл, додають 5 мл розчину
натрію гідроксиду. Колбу накривають вологим червоним лакмусовим папірцем
та годинниковим склом і нагрівають на водяному обігрівачі протягом 5 хвилин.
2.3.4. Дослідження на солі кальцію
Розчини солей кальцію з розчином амонію оксалату утворюють білий
дрібнокристалічний осад, нерозчинний в оцтовій кислоті, але легко розчинний
в хлороводневій та нітратній кислотах:
Реакцію проводять у середовищі аміачного буферу.
Гранична чутливість реакції – 0,0035 мг іона кальцію в 1 мл розчину.
Методика. До 10 мл розчину лікарського засобу, приготовленого за вимогами
монографії, додають 1 мл розчину амонію хлориду, 1 мл розчину амоніаку та
1 мл розчину амонію оксалату. Розчин перемішують і через 10 хвилин
порівнюють з еталоном, який складається з 10 мл еталонного розчину Б іона
кальцію і такої ж кількості реактивів, які додані до розчину лікарської
речовини, що досліджується.
2.3.5. Дослідження на солі феруму
Для визначення домішки іонів феруму, незалежно від ступеню
окиснення, застосовують розчин сульфосаліцилової кислоти, яка утворює з
ними в амоніачному середовищі жовто-коричневий ферилсульфосаліцилатний
комплекс:
Гранична чутливість реакції – 0,00005 мг іона феруму в 1 мл розчину.
NH4OH NH3 + H2O
NH4
+ + OH
-
NH4OH
Ca2+ + C2O4
2- CaC2O4
C O
O
O
S
O
O
OC O
O
O
S
O
O
O
3-
2
Fe
6-
Fe
3
6NH4 ;
+ 3NH4
+
64.
65
Методика. До 10мл розчину лікарського засобу, приготовленого за вимогами
монографії, додають 2 мл розчину сульфосаліцилової кислоти та 1 мл розчину
амоніаку і через 5 хвилин порівнюють з еталонним розчином Б іонів феруму
(10 мл), до якого додано таку ж кількість реактивів, як і до розчину, що
досліджується.
У сполуках магнію визначення домішки іонів феруму проводять у
середовищі амоніачного буферу, щоб запобігти утворенню осаду гідроксиду
магнію.
При визначенні домішки феруму в сполуках магнію поступають так само,
як зазначено вище, але перед додаванням розчину амоніаку до розчину
лікарського засобу додають 0,5 мл розчину амонію хлориду.
При визначенні домішок феруму у солях алюмінію застосовують розчин
натрію гідроксиду, в якому гідроксид алюмінію розчиняється.
Визначення солей феруму в сполуках алюмінію проводять таким чином:
до розчину лікарського засобу, приготовленого як зазначено у відповідній
монографії, додають 5 мл розчину сульфосаліцилової кислоти і 2 мл розчину
натрію гідроксиду і порівнюють з еталонним розчином Б іона феруму (10 мл),
до якого додають таку ж кількість реактивів, як і до розчину, що досліджується.
Визначення солей феруму в органічних сполуках проводять після
спалювання наважки лікарського засобу в концентрованій сульфатній кислоті.
Залишок після спалювання обробляють 2 мл концентрованої хлороводневої
кислоти при нагріванні на водяному обігрівачі і додають 2 мл води. Якщо
потрібно, одержаний розчин фільтрують, фільтр промивають 3 мл води. Розчин
разом з промивними водами нейтралізують концентрованим розчином
амоніаку, доводять об’єм розчину водою до 10 мл. Далі чинять, як зазначено
вище.
2.3.6. Дослідження на солі цинку
Домішку іонів цинку визначають по утворенню білого осаду або муті при
взаємодії з розчином калію гексаціаноферату (II):
Гранична чутливість реакції – 0,001 мг цинк-іона в 1 мл розчину.
Методика. До 10 мл розчину лікарського засобу, приготовленого за вимогами
монографії, додають 2 мл хлороводневої кислоти, 5 крапель розчину калію
фероціаніду і через 10 хвилин порівнюють з еталонним розчином Б іона цинку
(10 мл), до якого додають таку ж кількість реактивів, як і до розчину, що
досліджується.
Проведенню досліджень заважає наявність солей феруму (III). Його
вплив усувається додаванням гідроксиду амонію, котрий утворює з солями
феруму осад гідроксиду Fe(OH)3, який відфільтровують. Солі цинку
залишаються у фільтраті у вигляді розчинних цинкатів [Zn(OН)4]2-
.
У разі появи в розчині, що досліджується, синього забарвлення,
необхідно спочатку відділити ферум. Для цього до такого розчину, нагрітого до
3Zn2+ + 2K+ + 2[Fe(CN)6]4- K2Zn3[Fe(CN)6]2
65.
66
кипіння, додають розчинамоніаку до виразного запаху, суміш фільтрують. В
фільтраті визначають домішку цинку.
2.3.7. Дослідження на солі важких металів
При визначенні домішки солей важких металів як модельний іон
відкривають іон плюмбуму, оскільки він за хімічними властивостями дуже
близький до інших важких металів.
Розчини солей плюмбуму в залежності від концентрації дають з розчином
натрію сульфіду чорний осад або буре забарвлення розчину:
Реакцію проводять у середовищі оцтової кислоти.
Гранична чутливість реакції – 0,0005 мг плюмбум-іона в 1 мл розчину.
Методика. До 10 мл розчину лікарського засобу, приготовленого за вимогами
монографії, додають 1 мл розведеної оцтової кислоти, 2 краплі розчину натрію
сульфіду, перемішують і через 1 хвилину порівнюють з 1 мл еталонного
розчину іона плюмбуму, розбавленого 9 мл води, до якого додано таку ж
кількість реактивів, як і до розчину, що досліджується.
Спостереження забарвлення проводять по осі пробірок діаметром близько
1,5 см, розміщених над білою поверхнею.
У порівнюваних розчинах допустима лише слабка опалесценція від сірки,
що може виділитися з натрію сульфіду.
При визначенні важких металів у зольному залишку органічних
лікарських засобів залишок обробляють гарячим розчином амонію ацетату,
водою та фільтрують. Домішку важких металів визначають у фільтраті
звичайним методом. Ферум визначенню не заважає, оскільки основні ацетати
феруму, що утворилися, залишаються на фільтрі.
2.3.8. Дослідження на арсен
Якщо в монографії немає спеціальної вказівки, то дослідження треба
проводити за методом I.
Метод I. Сполуки арсену під дією цинку та хлороводневої або сульфатної
кислоти відновлюються до арсину, який з меркурію дихлоридом утворює
сполуки, забарвлені від жовтого до оранжевого кольору, а після обробки
розчином калію йодиду – в бурувато-коричневий колір:
-2 e
As2O3 + 6Zn + 6H2SO4 2AsH3 + 6ZnSO4 + 3H2O
As+3 As-3
Zn0 Zn2+
+6 e 1
3
3HgCl2 + AsH3 As(HgCl)3 + 3HCl
HgCl2 + AsH3 AsH2(HgCl) + HCl
2HgCl2 + AsH3 AsH(HgCl)2 + 2HCl
As(HgCl)3 + AsH3 As2Hg3 + 3HCl
Pb2+ + S2- PbS
66.
67
Мінімальна кількість арсену,яка може бути відкрита цим методом
становить 0,0005 мг.
Методика. У колбу, в якій знаходиться попередньо підготовлена лікарська
речовина, додають від 10 до 12 крапель розчину дихлориду олова, 2 г
гранульованого цинку (який не містить арсену) і відразу закривають колбу
пробкою зі вставленою в неї верхньою частиною приладу. Вміст колби
обережно збовтують і залишають на 1 годину. При цьому температура
реакційної суміші не повинна перевищувати 40оС.
Паралельно в другому такому ж приладі проводять контрольний дослід з
усіма реактивами та додаванням 0,5 мл еталонного розчину арсену. Через
1 годину смужку паперу, оброблену розчином меркурію дихлориду, вміщують
у розчин калію йодиду для більш чіткого проявлення забарвлення та видалення
надлишку меркурію дихлориду:
Через 10 хвилин розчин калію йодиду зливають, смужку паперу ретельно
промивають декілька разів водою методом декантації в тій же склянці та
висушують між листками фільтрувального паперу.
Метод II. Ґрунтується на відновленні сполук арсену до металічного
арсену. Як відновник застосовують розчини натрію гіпофосфіту та
хлороводневої кислоти:
При нагріванні металічний арсен, який утворюється, спостерігають у
вигляді бурого осаду або забарвлення:
Якщо до охолодженої реакційної суміші додати ефір та перемішати,
арсен збирається на межі двох рідин у вигляді бурої плівки.
Метод також застосовують для визначення селену (червоне забарвлення)
та телуру (коричневе забарвлення) або у випадках визначення домішки арсену в
лікарських речовинах, які мають у своєму складі бісмут, меркурій, аргентум,
сульфіди та сульфіти.
Гранична чутливість реакції – 0,01 мг арсену в 10 мл реакційної суміші.
Методика. Наважку лікарської речовини (підготовлену згідно з вимогами
монографії) вносять у пробірку, додають 5 мл розчину натрію гіпофосфіту,
вміщують у киплячий водяний обігрівач і нагрівають протягом 15 хвилин.
В розчині, що досліджується, не повинно спостерігатися ні побуріння, ні
утворення бурого осаду.
У разі побуріння або утворення бурого осаду в пробірку після
охолодження додають 3 мл води, 5 мл ефіру та ретельно збовтують. При
наявності арсену на межі рідин утворюється бура плівка.
HgCl2 + 2KI HgI2 + 2KCl
HgI2 + 2KI K2[HgI4]
NaH2PO2 + HCl H3PO2 + NaCl
As2O3 + 3H3PO2 2As + 3H3PO3
2AsCl3 + 3H3PO2 + 3H2O 2As + 3H3PO3 + 6HCl
67.
68
2.3.9. Визначення леткихречовин та води
Визначення летких речовин та води проводиться такими методами:
1. Термічний метод – метод висушування. Точну наважку речовини вміщують
у бюкс та висушують до постійної маси (умови висушування вказані у
відповідній статті). Постійна маса вважається досягнутою, якщо два
послідовних зважування після 1 години висушування дають різницю, яка не
перевищує 0,0005 г. Визначення вологи, якщо температуру не зазначено,
проводиться при 100-105оС.
2. Об’ємний метод (метод дистиляції) – метод Діна-Старка. Відгонку води
здійснюють за допомогою органічних розчинників (толуол, ксилол, бензол,
чотирихлористий вуглець), з якими вода дає азеотропні суміші. Потім
вимірюють об’єм води у відгоні.
3. Метод титрування реактивом К. Фішера. Реактив К. Фішера – це розчин
діоксиду сірки, йоду та піридину в метиловому спирті. Взаємодія реактиву з
водою проходить у два етапи:
Метод дозволяє визначити сумарний вміст як вільної, так і
кристалізаційної води в органічних та неорганічних лікарських речовинах.
Реактиви і розчини, які використовуються в цьому методі необхідно зберігати в
умовах, що запобігають поглинанню атмосферної вологи.
Визначення води за методом К. Фішера проводять таким чином: точну
наважку лікарського засобу, що містить приблизно від 0,03 до 0,05 г води,
вміщують у суху колбу ємністю 100 мл, в яку попередньо додають 5 мл
метилового спирту. Перемішують 1 хвилину і титрують реактивом К. Фішера,
додаючи його при наближенні до кінцевої точки по 0,1-0,05 мл.
Кінець титрування може бути визначений як візуально по зміні
забарвлення від жовтого до червоно-коричневого, так і електрометричним
титруванням “до повного припинення струму”.
Паралельно титрують 5 мл метилового спирту (контрольний дослід).
N
S
OO
O
N
H
N
S
OO
O
N
H
N
3 + I2 + SO2 + H2O +2 +
+ CH3OH + SO4CH3
-
I
-
68.
69
Вміст води увідсотках обчислюють за формулою:
в
Тба
X
100)(
%
⋅⋅−
= , (2.3)
де а – об’єм реактиву К. Фішера, витраченого на титрування в основному
досліді, мл;
б – об’єм реактиву К. Фішера, витраченого на титрування в контрольному
досліді, мл;
в – наважка лікарського засобу, г;
Т – титр реактиву К. Фішера.
69.
70
3. Методи кількісногохімічного аналізу лікарських
речовин
3.1. Гравіметричний аналіз
Гравіметричний аналіз базується на вимірюванні маси речовини відомого
складу, утвореної з речовини або вилученої з препарату які визначають.
Для кількісного визначення лікарських речовин найчастіше застосовують
метод преципітації (рідше – методи відгонки або екстракції), який ґрунтується
на осадженні речовини, що визначається, у вигляді малорозчинної сполуки
відомого складу з наступним фільтруванням, прожарюванням (або
висушуванням) та зважуванням отриманого продукту.
Гравіметричне визначення складається з двох експериментальних
вимірювань:
– зважування наважки;
– зважування продукту відомого складу, отриманого з цієї наважки (треба
пам’ятати, що висушування або прожарювання і зважування продукту
необхідно проводити до отримання постійної маси).
На базі цих даних можна розрахувати вміст речовини, що визначається, у
відсотках:
наважкимаса
сявизначаєтьщоречовинимаса
X
100,
%
⋅
= (3.1)
Частіше маса речовини, що визначається, безпосередньо не вимірюється.
Замість цього виділяють і зважують продукт її взаємодії з реагентом. Щоб
знайти масу речовини, необхідно масу продукту помножити на гравіметричний
фактор (F), який є відношенням еквівалентної маси речовини, що визначається,
до еквівалентної маси зважуваного продукту:
продуктузваженогомасаамолекулярнb
сявизначаєтьщоречовинимасаамолекулярнa
F
⋅
⋅
=
,
, (3.2)
де а та b – коефіцієнти, на які треба помножити молекулярні маси,
щоб числа молей в діленому та дільнику були хімічно еквівалентні.
Таким чином, у загальному вигляді формула розрахунку процентного
вмісту лікарської речовини буде мати вигляд:
наважкимаса
Fсявизначаєтьщоречовинимаса
X
100,
%
⋅⋅
= , (3.3)
Головною перевагою гравіметричного методу аналізу є його висока
точність. Хоча в кожному окремому випадку ступінь точності залежить від
багатьох факторів – розчинності осаду, спільного осадження та інше. Найбільш
високою точність гравіметричного методу є у випадку, коли вміст речовини, що
визначається, перевищує 1%. Помилка в цьому випадку в межах 0,2-0,4%.
70.
71
До недоліків методутреба віднести велику клопітність і тривалість
проведення аналізу (що пов’язано з необхідністю виконання таких операцій як
фільтрування, промивання осаду, висушування або доведення до постійної
маси), а також значно низьку селективність у випадку аналізу
багатокомпонентних сумішей, що погіршує точність визначення.
Гравіметрія, застосовується при аналізі таких лікарських речовин як
натрію сульфат, солі хініну, солі бензилпеніціліну, метиландростендіол у
таблетках, прогестерон, тіаміну бромід. У сучасній аналітичній НТД
гравіметрія використовується дуже рідко.
Визначення хініну гідрохлориду
Осаджують основу хініну за допомогою розчину натрію гідроксиду:
C20H24N2O2
. HCl + NaOH = ↓C20H24N2O2 + NaCl + H2O
Основу екстрагують за допомогою хлороформу; хлороформний шар
відділяють, хлороформ відганяють. Залишок висушують та зважують.
Розрахунок проводять за формулою 3.3. В даному випадку:
112,1
94,356
92,396
)(.
)(.
222420
222420
==
⋅
=
ONHCмасамол
HClONHСмасамол
F
Методика. Близько 0,5 г речовини(точну наважку) вміщують у ділильну лійку
ємністю 100 мл, розчиняють у 20 мл води, додають 5 мл розчину натрію
гідроксиду і виділену основу екстрагують хлороформом 1 раз 20 мл і 3 рази по
10 мл. Хлороформний екстракт переносять в іншу ділильну лійку, промивають
водою 2 рази по 10 мл. Дають рідині добре розшаруватися та обережно
зливають хлороформний шар крізь змочений хлороформом фільтр, що містить
1,5-2,0 г безводного натрію сульфату. Фільтр і натрію сульфат промивають
20 мл хлороформу, приєднуючи його до основного розчину. Хлороформ
відганяють на водяному обігрівачі, до залишку додають 2 мл абсолютного
спирту, спирт відганяють досуха на водяному обігрівачі. Залишок сушать при
100-105оС до постійної маси. Маса залишку помножена на 1,112 відповідає
кількості хініну гідрохлориду у взятій наважці.
3.2. Титриметричні (об’ємні) методи аналізу
Титриметричний (або об’ємний) аналіз базується на визначенні
кількісного вмісту речовини за кількістю використаного стандартного розчину.
Титриметричні методи застосовуються в фармацевтичному аналізі
найбільш широко, оскільки вони не потребують великих затрат часу, зручні та
забезпечують достатній ступінь точності.
Стандартні розчини, які застосовують для титрування, мають назву
титрованих. Найчастіше концентрацію титрованих розчинів виражають через:
71.
72
- Молярність (М)– виражена в молях кількість розчиненої речовини, що
міститься в одному літрі розчину. Молярність розраховують як відношення
кількості розчиненої речовини до об’єму розчину (розмірність – моль/л).
Згідно з діючою в Україні АНД за одиницю молярності приймають моль так
званих “умовних часток” речовини. Під “умовною часткою” розуміють
частку молекули, яка відповідає за передачу електрону або перенос однієї
одиниці заряду в перебігу окисно-відновних або об’ємних реакцій
відповідно. Тобто, фактично, термін “умовна частка” збігається з поняттям
“еквівалент”.
Слід зазначити, що в Європейській фармакопеї за одиницю молярності
титрованих розчинів прийнято моль молекул розчиненої речовини.
Для порівняння: один літр 0,01 моль/л розчину йоду згідно з діючою АНД
містить 1,269 г йоду (УЧ=1/2 I2), а за Європейською фармакопеєю в одному
літрі 0,01 моль/л розчину йоду міститься 2,54 г йоду.
- Титр – виражена в грамах маса розчиненої речовини, яка міститься в
одному мілілітрі розчину. Титр розраховують як відношення маси
розчиненої речовини до об’єму розчину (розмірність – г/мл).
Титр титранту за речовиною, що визначається, – це виражена в
грамах маса речовини, що визначається, яка реагує з 1 мл титрованого розчину
(розмірність – г/мл).
Титр за речовиною, що визначається, розраховують за формулою:
1000
EM
T
⋅
= , (3.4)
де М – молярність титрованого розчину, моль/л;
Е – молярна маса еквіваленту речовини, що визначається, г/моль.
Титровані розчини виготовляють з хімічно чистих речовин. Від точності
концентрації титрованого розчину залежить точність визначення.
У випадках, коли концентрація виготовленого розчину відрізняється від
теоретичної (внаслідок складності виготовлення або змін у результаті
зберігання), розраховують коефіцієнт поправки до молярності.
Коефіцієнт поправки (К) показує, у скільки разів концентрація
виготовленого розчину відрізняється від теоретичної. Допускається коефіцієнт
поправки у межах від 0,98 до 1,02.
Титровані розчини зручно виготовляти розчиненням в необхідному
об’ємі фіксаналів – запаяних ампул, в яких містяться речовини в точно
визначеній кількості.
Головною умовою точності титриметричного визначення є додавання
титрованого розчину в кількості, хімічно еквівалентній кількості речовини, що
визначається. Момент титрування, в який досягається ця умова, називається
точкою еквівалентності. Щоб на практиці визначити точку еквівалентності,
необхідно зафіксувати зміну якої-небудь фізичної властивості (забарвлення
розчину, електродний потенціал, електропроводність та ін.) системи в цій точці
або поблизу неї. Точка, в якій ці зміни стають помітними, має назву кінцевої
точки титрування.
72.
73
Між кінцевою точкоютитрування та точкою еквівалентності завжди є
деяка різниця, зумовлена неадекватністю зміни фізичної властивості та
здатністю дослідника фіксувати цю зміну.
Частіше за все кінцеву точку титрування фіксують за зміною забарвлення
розчину або індикатору.
За способом проведення розрізняють методи прямого, зворотного і
непрямого (посереднього, замісникового) титрування.
Пряме титрування базується на безпосередньому вимірюванні об’єму
титрованого розчину, витраченого на взаємодію з речовиною, що визначається.
Розрахунок проводять за формулою:
m
TKV
X
100
%
⋅⋅⋅
= , (3.5)
де V – об’єм титранту, витрачений на титрування, мл;
К – коефіцієнт поправки;
Т – титр титрованого розчину за речовиною, що визначається, г/мл;
m – маса наважки речовини, що визначається, г.
Зворотне титрування застосовують, коли реакція між речовиною, що
визначається, та титрованим розчином проходить повільно, однак, до кінця;
коли визначають леткі речовини та в деяких інших випадках. При зворотному
титруванні вимірюють два об’єми: об’єм титрованого розчину I, який реагує з
речовиною, що визначається, і додається в надлишку, та об’єм титрованого
розчину II, яким надлишок розчину I відтитровують.
Розрахунок вмісту речовини проводять за різницею між об’ємами:
m
TKVKV
X
100)(
% 2211 ⋅⋅⋅−⋅
= , (3.6)
де V1 – об’єм титрованого розчину I, мл;
V2 – об’єм титрованого розчину II, мл;
К1, К2 – коефіцієнти поправки;
Т – титр розчину I за речовиною, що визначається, г/мл;
m – маса наважки речовини, що визначається, г.
Непрямі (посередні) методи титрування (або титрування за замісником)
застосовують для речовин, які не можуть кількісно прореагувати з титрованим
розчином. При непрямих методах титрування відтитровують продукт, який
виділяється в еквівалентній кількості при взаємодії речовини, що визначається,
з якою-небудь третьою речовиною. Результат непрямого титрування так само,
як і прямого, розраховують за формулою 3.5.
В окремих випадках при виконанні титриметричного визначення
необхідно проведення контрольного досліду. Якщо в методиці немає особливих
вказівок, контрольний дослід полягає в точному відтворенні методики, але без
додавання речовиною, що визначається. Контрольний дослід необхідний для
одержання більш точних результатів при визначеннях, пов’язаних з реакціями,
які перебігають повільно (частіше при зворотному титруванні), при
73.
74
застосуванні стандартних розчинівсильних окисників, летких речовин та в
деяких інших випадках.
Об’єм титрованого розчину, який прореагував з речовиною
розраховують:
а) при прямому титруванні за різницею (V – Vк);
б) при зворотному титруванні за різницею (Vк – V),
де V – об’єм титрованого розчину, витраченого в основному досліді;
Vк – об’єм титрованого розчину, витраченого в контрольному досліді.
3.2.1. Методи осадження. Аргентометрія
Методи осадження базуються на утворенні при титруванні
малорозчинних речовин, які випадають в осад. Для кількісних розрахунків за
цими методами необхідно визначити об’єм титранту, який витрачається на
повне осадження речовиною, що визначається.
В аналітичній хімії відомо багато реакцій, які супроводжуються
утворенням осадів. Однак, на практиці можуть застосовуватися тільки ті з них,
які відповідають таким вимогам:
1. Осад повинен бути практично нерозчинним (добуток розчинності ДР=10-9
).
2. Утворення осаду повинно відбуватися швидко.
3. Реакції осадження повинні перебігати кількісно у відповідності зі
стехіометрією хімічного рівняння.
4. Повинна бути можливість вибору індикатору до відповідної реакції
осадження.
5. Результати титрування не повинні помітно спотворюватися явищами
адсорбції. Поблизу точки еквівалентності допускається повільне додавання
титранту та інтенсивне перемішування для усунення впливу адсорбції.
Найбільшою мірою цим умовам відповідають реакції утворення осадів
ряду аніонів з солями аргентуму, на яких базується аргентометрія. За цим
методом визначають хлорид-, бромід-, йодид-, тіоціанат-іони.
Визначення точки еквівалентності проводиться візуально за допомогою
індикаторів або потенціометрично. В аргентометрії застосовуються індикатори,
які утворюють забарвлений осад, забарвлений комплекс або ж адсорбційні
індикатори.
В залежності від того, який індикатор використовується, аргентометричне
титрування поділяють на декілька методів, основними з яких є методи Мора,
Фольгарда і Фаянса-Ходакова.
3.2.1.1. Метод Мора дозволяє кількісно визначати хлориди та броміди.
Індикатором є калію хромат, який з надлишковою краплею аргентуму нітрату
утворює цегельно-червоний осад аргентуму хромату. Застосування калію
хромату як індикатора у цьому методі ґрунтується на тому, що розчинність
аргентуму хромату значно вища від розчинності аргентуму хлориду чи броміду.
74.
75
Тому спочатку відбуваєтьсявипадіння аргентуму броміду і хлориду, а вже
після їх повного осадження – хромату:
Умови титрування:
1. Середовище близьке до нейтрального (pH 6,3-10,0). У кислому середовищі
індикатор перетворюється у дихромат:
Дихромат-іон не може бути індикатором через високу розчинність Ag2Cr2O7.
При pH>10,0 можливе протікання реакції:
2. Поблизу точки еквівалентності необхідно титрувати повільно при сильному
перемішуванні для посилення десорбції галогенід-іонів з поверхні осаду.
Метод Мора не дозволяє визначити йодид-іони внаслідок сильної
адсорбції індикатора на поверхні осаду аргентуму йодиду. Забарвлення
з’являється до настання моменту еквівалентності й сам момент еквівалентності
спостерігається не чітко.
Визначення натрію (калію) хлориду
Методика. Близько 1 г речовини (точна наважка) розчиняють у воді в мірній
колбі ємністю 50 мл та доводять об’єм розчину до мітки; 5 мл одержаного
розчину розбавляють водою до об’єму 40 мл і титрують 0,2 моль/л розчином
аргентуму нітрату до оранжево-жовтого забарвлення (індикатор – калію
хромат).
Е=М.м.; 1 мл 0,1 моль/л розчину аргентуму нітрату відповідає 0,007456 г
калію хлориду і 0,005844 г натрію хлориду, яких повинно бути не менш 99,5%.
Розрахунок кількісного вмісту натрію (калію) хлориду у відсотках
проводять за формулою:
п
МК
Vm
VTKV
X
⋅
⋅⋅⋅⋅
=
100
% , (3.7)
де V – об’єм 0,1 моль/л розчину аргентуму нітрату, витраченого на
титрування наважки натрію (калію) хлориду, мл;
К – коефіцієнт поправки до молярності 0,1 моль/л розчину
аргентуму нітрату;
Т – титр 0,1 моль/л розчину аргентуму нітрату за натрію (калію)
хлоридом, г/мл;
Vмк – об’єм мірної колби, мл;
Vп – об’єм піпетки, мл;
m – маса наважки натрію (калію) хлориду, г.
2Ag+ + CrO4
2- Ag2CrO4
Ag+ + Cl
-
AgCl
2CrO4
2- + 2H+ Cr2O7
2- + H2O
2Ag+ + 2OH
-
2AgOH Ag2O + H2O
75.
76
3.2.1.2. Метод Фольгардабазується на осадженні хлоридів, бромідів, йодидів
надлишком стандартного розчину аргентуму нітрату з наступним його
відтитровуванням стандартним розчином роданіду (тіоціанату) амонію.
Як індикатор в методі Фольгарда використовують іон феруму(III), який
вводиться у розчин у вигляді залізо-амонієвого галуну.
Застосування феруму(III) як індикатора базується на його здатності
утворювати з роданід-іонами у водних розчинах комплексну сполуку криваво-
червоного кольору:
Іони аргентуму утворюють з роданід-іонами важкорозчинну сполуку:
Розрахунки показують, що утворення комплексу феруму з роданід-іонами
почнеться тільки після повного осадження катіонів аргентуму у вигляді
аргентуму роданіду, а потім зайва крапля розчину амонію роданіду буде
реагувати з іоном феруму(III), забарвлюючи розчин у червоний колір.
Умови титрування:
1. Середовище повинно бути кислим. Це необхідно для пригнічення гідролізу
іону феруму(III), оскільки індикатор – це сіль, утворена слабкою основою і
сильною кислотою:
Продукт гідролізу – гідроксид феруму(III) червоно-бурого кольору –
заважає точному встановленню точки еквівалентності. Для пригнічення
гідролізу розчини підкислюють нітратною кислотою.
2. При титруванні іонів аргентуму роданідом амонію перша зміна забарвлення
розчину відбувається приблизно за 1% до моменту еквівалентності, що
пов’язано з адсорбцією осадом аргентуму роданіду іонів аргентуму. Для
прискорення їх десорбції з поверхні осаду необхідно енергійне перемішування
розчину у кінці титрування.
З метою економії розчину аргентуму нітрату застосовують непрямий
метод Фольгарда. Точну наважку солі галогеніда розчиняють у воді,
підкислюють нітратною кислотою, додають 1мл розчину залізо-амонієвого
галуну і 0,1 мл 0,1 моль/л розчину амонію роданіду. Виникає криваво-червоне
забарвлення:
FeNH4(SO4)2 + 3NH4SCN → [Fe(SCN)3] + 2(NH4)2SO4
Розчин титрують 0,1 моль/л розчином аргентуму нітрату. Спочатку
реагує галогенід, а після досягнення моменту еквівалентності надлишкова
крапля титранту реагує з феруму (ІІІ) роданідом внаслідок чого розчин
знебарвлюється:
NaBr +AgNO3 → AgBr↓ + NaNO3
3AgNO3 + [Fe(SCN)3] → 3AgSCN↓ + Fe(NO3)3
NH4
+
+ Fe
3+
+ 2SO4
2-
Fe3+
+3H2O Fe(OH)3 + 3H+
FeNH4(SO4)2
Fe3+ + 3SCN
-
[Fe(SCN)3]
Ag+ + SCN
- AgSCN
76.
77
Розрахунок кількісного вмістугалогеніду у відсотках проводять за
формулою 3.6, віднімаючи від об’єму аргентуму нітрату, який пішов на
титрування, доданий об’єм амонію роданіду.
Визначення бромізовалу
Оскільки атом брому в молекулі бромізовалу ковалентно зв’язаний, його
попередньо переводять в іонний стан кип’ятінням з лугом:
Потім зворотнім титруванням визначають натрію бромід:
Е=М.м.
Методика. Близько 0,3 г субстанції (точна наважка) вміщують у конічну колбу
ємністю 250 мл, додають 20 мл 1 моль/л розчину натрію гідроксиду та
нагрівають на киплячому водяному обігрівачі зі зворотнім холодильником
протягом 15 хв. Після охолодження розбавляють 50 мл води, додають 15 мл
розведеної нітратної кислоти, 25 мл 0,1 моль/л розчину аргентуму нітрату,
енергійно перемішують та надлишок аргентуму нітрату відтитровують
0,1 моль/л розчином амонію роданіду (індикатор – залізо-амонієвий галун).
Паралельно проводять контрольний дослід.
1 мл 0,1 моль/л розчину аргентуму нітрату відповідає 0,02231 г
бромізовалу, якого в субстанції має бути не менш 97%.
Розрахунок кількісного вмісту бромізовалу у відсотках проводять за
формулою:
m
TKVV
X к 100)(
%
⋅⋅⋅−
= , (3.8)
де Vк – об’єм 0,1 моль/л розчину амонію роданіду, витраченого на
титрування у контрольному досліді, мл;
V – об’єм 0,1 моль/л розчину амонію роданіду, витраченого на
титрування у досліді з наважкою, мл;
К – коефіцієнт поправки до молярності 0,1 моль/л розчину амонію
роданіду;
Т – титр 0,1 моль/л розчину аргентуму нітрату за бромізовалом, г/мл;
m – маса наважки бромізовалу, г.
C CH CO NH C NH2
H3C
H3C
H Br O
+ 4NaOH
t
C CH
H3C
H3C
H OH
COONa + 2NH3 + Na2CO3 + NaBr
NaBr + AgNO3 AgBr + NaNO3
AgNO3 + NH4SCN AgSCN + NH4NO3
FeNH4(SO4)2 + 3NH4SCN Fe(SCN)3 + 2(NH4)2SO4
77.
78
3.2.1.3. Метод Фаянсапередбачає використання для аргентометричного
титрування адсорбційних індикаторів при визначенні хлорид-, бромід- та
йодид-іонів.
Адсорбційні індикатори – це слабкі органічні кислоти, які дисоціюють за
схемою:
Найчастіше як адсорбційні індикатори використовують флуоресцеїн, еозин,
еозинат натрію, бромфеноловий синій та ін.
У процесі титрування галогенід-іонів іонами аргентуму формуються
осади галогенідів аргентуму, які схильні до утворення колоїдів. Поки не
досягнуто моменту еквівалентності, колоїдні частинки галогенідів аргентуму
адсорбують галогенід-іони, які є у надлишку, і набувають негативного заряду.
Негативно заряджені частинки осаду притягують до себе іони протилежного
заряду, наприклад:
В момент еквівалентності, який співпадає з ізоелектричною точкою, осад
не буде мати заряду.
Перша ж надлишкова краплина розчину аргентуму нітрату створює у
розчині надлишок іонів аргентуму, які починають адсорбуватися на осаді
йодиду аргентуму, надаючи осаду позитивного заряду: ↓[(AgI)Ag+
]NO3
-. Як
протийони поряд з нітрат-іонами будуть також адсорбуватися забарвлені аніони
індикатору: ↓[(AgI)Ag+
]Ind-. Це призведе до зміни забарвлення поверхні осаду.
Зміна забарвлення поверхні осаду свідчить про необхідність припинити
титрування.
Умови титрування:
1. Титрування ведуть при значенні pH, яке визначено для кожного
адсорбційного індикатора, наприклад: з флуоресцеїном в нейтральному або
слабокислому середовищі, з еозином – в кислому, при pH 2.
2. Оскільки адсорбція іонів індикатору відбувається на поверхні осаду, то
вигідно, щоб вона була якомога більшою. Особливо розгалужену поверхню
мають колоїдні розчини. Тому титрування з адсорбційними індикаторами
краще вести, якщо осад хоча б частково знаходиться у вигляді колоїдних
часток.
Щоб запобігти коагуляції колоїдного розчину, до розчину додають
захисні колоїди (антикоагулянти): декстрин або крохмаль.
Методом Фаянса з бромфеноловим синім визначають кількісний вміст
хлоридів, бромідів і йодидів. При спільній присутності титрується вся сума
галогенідів.
Методом Фаянса з еозинатом натрію визначають йодиди. Визначенню не
заважають хлориди, заважають броміди.
H
+
+ Ind
-
HInd
AgI + KNO3AgNO3 + KI [(AgI)I ]K+
78.
79
Визначення натрію (калію)йодиду
Методика. Близько 0,3 г субстанції (точна наважка), попередньо висушеної
при 110оС протягом 4 год, розчиняють у 30 мл води, додають 1,5 мл розведеної
оцтової кислоти, 5 крапель 0,1% розчину еозинату натрію і титрують 0,1 моль/л
розчином аргентуму нітрату до переходу забарвлення осаду від жовтого до
рожевого.
Е=М.м.; 1 мл 0,1 моль/л розчину аргентуму нітрату відповідає 0,01660 г
калію йодиду, якого у висушеній субстанції повинно бути не менш 99,5%, або
0,01499 г натрію йодиду, котрого у субстанції повинно бути не менш 99,0%.
Розрахунок кількісного вмісту натрію (калію) йодиду у відсотках
проводять за формулою 3.5.
3.2.1.4. Метод Кольтгофа. На практиці досить часто доводиться стикатися з
необхідністю визначати одні галогеніди в присутності інших. Метод Кольтгофа
дозволяє визначити йодиди в присутності хлоридів і бромідів. Відомо, що
крохмаль утворює комплекс з йодом, забарвлений в синій колір лише в
присутності йодид-іонів. При зникненні з розчину йодид-іонів відбувається
руйнування комплексу і знебарвлення.
Методика. До розчину натрію або калію йодиду додають 20 – 30 мл води, 1
краплю 0,1 моль/л розчину калію йодату, 2 мл розчину крохмалю і по краплям
розведену сульфатну кислоту до появи синього забарвлення:
KIO3 + 5KI + 3H2SO4 → 3I2 + 3K2SO4 + 3H2O
Розчин, що досліджується титрують 0,1 моль/л розчином аргентуму
нітрату до зникнення синього забарвлення:
KI + AgNO3 → AgI↓ + KNO3
Примітки. Визначенню йодидів за методом Кольтгофа не заважають
хлориди. При наявності бромідів необхідно перед додаванням сульфатної
кислоти додати до розчину 5 мл 10% розчину карбонату амонію.
Визначення точки еквівалентності при аргентометричному титруванні
можна проводити також різними інструментальними методами, в тому числі
потенціометрично. У цьому випадку у розчин, що титрують, занурюють
електрод, потенціал якого визначається концентрацією або іонів аргентуму, або
іонів галогеніду. У точці еквівалентності відбувається різка зміна концентрації
цих іонів, і відповідно, потенціалу електроду, що свідчить про закінчення
титрування.
Таким чином, електрод виконує функцію індикатору, тому і називається
індикаторним.
3.2.2. Комплексонометрія
Комплексонометрія – титриметричний метод, який базується на реакціях
комплексоутворення іонів металів з комплексонами. Комплексонами називають
поліамінополікарбонові кислоти та їх солі, які належать до полідентатних
хелатоутворюючих сполук.
79.
80
Комплексони здатні утворюватиз дво-, три-, чотиривалентними металами
незалежно від їх валентності, в простому стехіометричному співвідношенні 1:1
стійкі, добре розчинні у воді комплексні сполуки.
Найчастіше для титрування застосовують динатрієву сіль
етилендіамінтетраоцтової кислоти (ЕДТА, трилон Б, комплексон III).
Точку еквівалентності у комплексонометричному титруванні можна
встановлювати за допомогою фізичних методів (потенціометричний,
амперометричний та ін.), однак на практиці віддають перевагу візуальному
індикаторному способу, як найбільш простому, зручному та швидкому.
Індикатори, які використовують в комплексонометрії, називають
металоіндикаторами. Металоіндикатори – це органічні барвники, які
утворюють з іонами металів інтенсивно забарвлені комплекси, колір яких
відрізняється від забарвлення вільного індикатору. Треба звернути увагу, що
більшість індикаторів здатні приєднувати або віддавати протони, змінюючи при
цьому забарвлення. Тому металохромні індикатори, є водночас pH-
індикаторами. В зв’язку з цим застосування їх у комплексонометрії можливе
тільки при певному значенні pH (в тій області, де конкуруюча реакція з
протонами відсутня).
Однією з умов комплексонометрї є вимога, щоб комплекс металу з
індикатором був менш міцним, ніж з трилоном Б.
До числа металоіндикаторів, які найчастіше застосовуються в аналітичній
практиці, можна віднести кислотний хром темно-синій, кислотний хром
чорний, пірокатехіновий фіолетовий, ксиленовий оранжевий та ін. Більшість з
металоіндикаторів в розчинах нестійкі й зберігаються тільки протягом
декількох днів. Застосовуються суміші їх з сухим хлоридом натрію або калію у
співвідношенні 1:200. Така суміш стійка тривалий час. Для титрування
застосовують 20-30 мг приготованої суміші на 100 мл розчину, що титрується.
Під час комплексонометричного визначення до розчину, який містить
катіон, що визначається, при суворому дотриманні відповідного значення pH
додають невелику кількість потрібного індикатору. Утворюється порівняно
стійка, добре розчинна у воді забарвлена сполука:
При титруванні трилоном Б спочатку утворюється комплекс з вільними
іонами металу, що визначається:
Коли всі вільні йони металу відтитровано, починається руйнування
комплексу індикатору з металом – починається перехід забарвлення. В момент
еквівалентності відбувається повне руйнування забарвленого
Me2+ + H2Ind MeInd + 2H+
+ 2H
+
CH2 N
CH2COONa
CH2COONa
NCH2
Me
CH2COO
CH2COO
Me2+
CH2 N
CH2COONa
CH2COOH
CH2COONa
CH2COOH
NCH2
+
80.
81
металоіндикаторного комплексу, індикаторзвільняється і розчин набуває
кольору вільного індикатору:
Пряме титрування застосовують для визначення іонів: Ca2+
, Mg2+
, Zn2+
, Bi3+
,
Co2+
, Mn2+
та ін.
При зворотному титруванні надлишок трилону Б, який не вступив у
реакцію з визначаємим металом, відтитровують при необхідному значенні pH з
відповідним індикатором розчином солі цинку, магнію або ін. Способом
зворотного титрування визначають іони Pb2+
, Hg2+
, As3+
та ін.
Визначення кальцію хлориду
Методика. Близько 0,8 г субстанції (точна наважка), зваженої в закритому
бюксі, розчиняють у воді, переносять у мірну колбу ємністю 100 мл, доводять
об’єм розчину водою до мітки та старанно перемішують. До 25 мл
приготованого розчину додають 5 мл амоніачного буферного розчину (pH 9,5-
10,0), 7 крапель розчину кислотного хром темно-синього (або 20-30 мг сухої
індикаторної суміші) та титрують 0,05 моль/л розчином трилону Б від
червоного до синьо-фіолетового забарвлення.
Е=М.м.; 1 мл 0,05моль/л розчину трилону Б відповідає 0,01095 г кальцію
хлориду, кількісний вміст якого в субстанції має бути не менш 98,0%.
В процесі титрування відбуваються такі реакції:
+ H2Ind+ MeInd
CH2 N
CH2COONa
CH2COONa
NCH2
Me
CH2COO
CH2COO
CH2 N
CH2COONa
CH2COOH
CH2COONa
CH2COOH
NCH2
+ 2H+
CH2 N
CH2COONa
CH2COONa
NCH2
Ca
CH2COO
CH2COO
+ Ca2+
CH2 N
CH2COONa
CH2COOH
CH2COONa
CH2COOH
NCH2
H+ + NH4OH NH4
+ + H2O
+ Ca2+
(червоного кольру)
Cl Cl
+ 2H+
комплекс індикатору з Ca2+
H2O OH2
Cl
Cl
N N
OH OH NHCOCH3
SO3Na
Cl
Cl SO3Na
O Ca O
NN
NHCOCH3
81.
82
Розрахунок кількісного вмістукальцію хлориду проводять за
формулою 3.7.
Визначення плюмбуму (ІІ) оксиду
Методика. Близько 0,2 г субстанції (точна наважка) розчиняють у 5 мл
концентрованої нітратної кислоти. До розчину додають 50 мл 0,05 моль/л
розчину трилону Б, 10 мл амоніачного буферного розчину і 0,1 г індикаторної
суміші кислотного хром чорного спеціального, перемішують до розчинення і
титрують 0,05 моль/л розчином сульфату цинку до переходу синього
забарвлення у червоне.
Е=М.м.; 1 мл 0,05моль/л розчину трилону Б відповідає 0,01116 г
плюмбуму оксиду, кількісний вміст якого в субстанції не менш як 99,0%.
В процесі титрування відбуваються такі реакції:
CH2 N
CH2COONa
CH2COONa
NCH2
Pb
CH2COO
CH2COO
CH2 N
CH2COONa
CH2COOH
CH2COONa
CH2COOH
NCH2
+ Pb2+ + 2H+
+ Zn2+ + 2H+
CH2 N
CH2COONa
CH2COOH
CH2COONa
CH2COOH
NCH2
CH2 N
CH2COONa
CH2COONa
NCH2
Zn
CH2COO
CH2COO
(надлишок)
+
CH2 N
CH2COONa
CH2COONa
NCH2
Ca
CH2COO
CH2COO
CH2 N
CH2COONa
CH2COOH
CH2COONa
CH2COOH
NCH2
+
Cl
H2O OH2
Cl
Cl SO3Na
O Ca O
NN
NHCOCH3
вільний індикатор (синьо-фіолетового кольору)
+
Cl
Cl
Cl
OH OH NHCOCH3
SO3Na
N N
82.
83
Розрахунок кількісного вміступлюмбуму (ІІ) оксиду у відсотках
проводять за формулою 3.6.
3.2.3. Меркуріметрія
Меркуріметрія належить до методів комплексонометричного титрування
з використанням неорганічних лігандів. Метод ґрунтується на утворенні
малодисоційованих сполук з катіоном меркурію (II) (Hg2+
).
Меркуріметрія дозволяє визначати лікарські речовини, які містять
галогенід-іони.
Як стандартні розчини, застосовують розчини меркурію (II) нітрату –
Hg(NO3)2 або меркурію (II) перхлорату – Hg(ClO4)2. При цьому відбуваються
такі реакції:
При кількісному визначенні хлоридів та бромідів титруванням розчинами
солей меркурію (II) як індикатори використовують натрію нітропрусид або
дифенілкарбазон.
Натрію нітропрусид [натрій пентаціанід нітрозил феррат (III)] з катіонами
меркурію (II) утворює білий осад:
При використанні дифенілкарбазону забарвлення розчину змінюється від
жовто-червоного до рожево-фіолетового:
Під час визначення йодидів у процесі титрування утворюється
безбарвний комплекс калію тетрайодмеркурату K2HgI4:
N N
C
NHN
C6H5
C6H5C6H5
C6H5
C
NH N
Hg
NN
OO2O C
NH NH
N N
C6H5
C6H5
+ Hg2+
2H+
Сумарно: 4KI + Hg(NO3)2 K2[HgI4] + 2KNO3
2KI + Hg(NO3)2 HgI2 + 2KNO3
HgI2 + 2KI K2HgI4
Na2[Fe(CN)5NO] 2Na+ + [Fe(CN)5NO]2-
Hg[Fe(CN)5NO]Hg2+ + [Fe(CN)5NO]2-
Cl
-
+ Hg2+ [HgCl]+
[HgCl]+ + Cl
-
HgCl2
OH2H2O
+ Zn2+ + 2H+
комплекс індикатору з Zn2+ (червоного кольору)вільний індикатор (синього кольору)
OH
NaO3S
O2N
OH
N N
O2N
NaO3S
O Zn O
NN
83.
84
Надлишкова крапля титрантуреагує з комплексом – комплекс руйнується
і випадає осад меркурію (II) йодиду червоного кольору; таким чином
використання індикатору при титруванні йодидів не потрібно:
Молярна маса еквіваленту в цьому випадку дорівнює двом молекулярним
масам: Е = 2М.м.
Переваги меркуріметричного методу:
1. Титрування галогенідів у кислому середовищі.
2. Багато йонів, які заважають визначенню при використанні методів Мора і
Фольгарда, у даному випадку не впливають на результат аналізу.
3. Не застосовуються дорогі та дефіцитні солі аргентуму.
4. Сполуки меркурію (II) легко регенеруються.
3.2.4. Кислотно-основне титрування у водному середовищі
Згідно з найбільш визнаною теорією кислот і основ – протолітичною
теорією Бренстеда і Лоурі – кислотно-основні реакції здійснюються за рахунок
переносу протону від кислоти до основи. Інакше кажучи, кислота є донором, а
основа – акцептором протонів.
Кислота і основа, які відрізняються вмістом протону, називаються
супряженими, наприклад: H2O і OH-; NH4
+
і NH3; CH3COOH і CH3COO-.
Метод кислотно-основного титрування ґрунтується на реакціях кислотно-
основної взаємодії, які в загальному вигляді можна представити таким чином:
Кислоти (нейтральні молекули, катіони або аніони) можуть віддавати
протон розчиннику або протонкоординуючий основі, при цьому в водних
розчинах утворюється іон гідроксонію H3O+
або в загальному випадку онієвий
іон. Чим дужчою є донорна кислота, тим слабша відповідна акцепторна основа.
Сила кислоти або основи значною мірою залежить від кислотно-основних
властивостей розчинника. В будь-якому розчиннику найдужчою кислотою є
сольватований протон – іон ліолію, а найдужчою основою – іон ліата (аніон
розчинника). Так, в водному розчині найдужчою кислотою є іон гідроксонію
H3O+
, а найдужчою основою – іон гідроксилу OH-; в рідкому амоніаку
найдужча кислота – іон амонію NH4
+
, а найдужча основа – амід-іон NH2
-; в
льодяній оцтовій кислоті найдужча основа – ацетат-іон CH3COO-, а найдужча
кислота – іон ацетонію CH3COOH2
+
.
Розчинення багатьох речовин можна представити як реакцію утворення
супряжених кислоти і основи:
K2[HgI4] + Hg(NO3)2 2HgI2 + 2KNO3
HA + B HB+ + A
-
кислота
кислота
супряжена супряженаоснова
основа
84.
85
Сила кислоти вводному розчині визначається тим, наскільки повно вона
віддає протони молекулам води, а сила основи – наскільки повно вона акцептує
протони молекул води.
Реакція між хлороводневою кислотою та водою протікає практично до
кінця, тому хлороводнева кислота належить до класу сильних кислот.
Сильними кислотами в водних розчинах є хлорна, хлороводнева, бромоводнева,
йодоводнева, сульфатна та нітратна кислоти.
Оцтова кислота реагує з водою меншою мірою, тому оцтова кислота
вважається слабкою кислотою.
Сила кислоти або основи оцінюється константою дисоціації, яка є
окремим випадком константи рівноваги, та виражається для кислот рівнянням:
а для основ:
Чим більшою є константа дисоціації, тим більша сила кислоти або
основи.
У випадку водних розчинів реакцію нейтралізації можна представити
таким чином:
При титруванні розчинів сильних кислот сильними основами і сильних
основ сильними кислотами у точці еквівалентності середовище нейтральне.
Після досягнення точки еквівалентності спостерігається різка зміна (стрибок)
pH, яку можна зафіксувати або інструментальними методами (наприклад,
потенціометричним), або візуально за допомогою кислотно-основних
індикаторів.
Кислотно-основні індикатори за своєю природою, як правило, слабкі
органічні кислоти або основи; при відщепленні або приєднанні протону
відбувається зміна їх забарвлення, що дає змогу використовувати ці сполуки
для фіксування кінцевої точки титрування. Найчастіше для аналізу лікарських
речовин застосовують такі кислотно-основні індикатори (табл. 3.1):
[B]
[BH+] [OH-
]
Kb =
H3O+ + OH
-
2H2O
CH3COOH + H2O CH3COO-
+ H3O+
NH3 + H2O NH4
+ + OH
-
HCl + H2O Cl
-
+ H3O+
[HA]
[H3O+] [A-
]
Ka = ,
85.
86
Таблиця 3.1
Інтервали рНі зміна кольору індикаторів
Назва
Інтервал рН
переходу
кольру
Зміна кольру
Метиловий фіолетовий 0,1 – 1,5 Жовтий – зелений
Малахітовий зелений 0,1 – 2,0 Жовтий – зеленкувато-блакитний
Крезоловий червоний 0,2 – 1,8 Червоний – жовтий
Крезоловий пурпуровий 1,2 – 2,8 Рожево-червоний – жовтий
Тимоловий синій 1,2 – 2,8 Червоний – жовтий
Тропеолін 00 1,4 – 3,2 Червоний – жовтий
Метиловий фіолетовий 1,5 – 3,2 Зелений – фіолетовий
Диметиловий жовтий 3,0 – 4,0 Червоний – жовтий
Метиловий оранжевий 3,0 – 4,4 Червоний – жовтий
Бромфеноловий синій 3,0 – 4,6 Жовтий – синій
Конго червоний 3,0 – 5,2 Синьо-фіолетовий – червоний
Бромкрезоловий зелений (синій) 3,8 – 5,4 Жовтий – синій
Алізариновий червоний С 4,6 – 6,0 Жовтий – пурпурово-червоний
Метиловий червоний 4,2 – 6.2 Червоний – жовтий
Лакмоїд 4,4 – 6,2 Червоний – синій
Бромкрезоловий пурпуровий 5,2 – 6,8 Жовтий – пурпуровий
Бромтимоловий синій 6,0 – 7,6 Жовтий – синій
Нейтральний червоний 6,8 – 8,0 Червоний – жовтий
Феноловий червоний 6,8 – 8,4 Жовтий – червоний
Крезоловий червоний 7,2 – 8,8 Жовтий – пурпурово-червоний
α-Нафтолфталеїн 7,4 – 6,8 Жовтувато-рожевий – зеленкувато-
синій
Крезоловий пурпуровий 7,4 – 9,0 Жовтий – фіолетовий
Тимоловий синій 8,0 – 9,6 Жовтий – синій
Фенолфталеїн 8,2 – 10,0 Безбарвний – яскраво-рожевий
Тимолфталеїн 9,4 – 10,6 Безбарвний – синій
Алізариновий жовтий Р 10,0-12,0 Світло-жовтий – червоно-оранжевий
Малахітовий зелений 11,4 – 13,0 Зеленкувато-блакитний – безбарвний
Індігокармін 11,6 – 14,0 Синій - жовтий
Різниця в інтервалах pH переходу забарвлення різних індикаторів
дозволяє для кожного конкретного випадку підібрати індикатор таким чином,
щоб інтервал переходу забарвлення індикатора знаходився в межах значень pH,
які співпадають зі стрибком кривої титрування.
При цьому слід пам’ятати, що кислотно-основне титрування сильно
розведених розчинів призводить до значного зменшення стрибка титрування і,
як наслідок, ускладнює фіксацію точки еквівалентності.
З тієї ж причини знижується точність визначення при титруванні слабких
кислот або основ. При Ка (Кв)≥10-8
зміни pH в кінцевій точці титрування
настільки малі, що зафіксувати їх з достатньою точністю неможливо.
86.
87
У ряді випадківдля більш чіткого визначення кінця титрування
застосовують контрольний розчин, який має у своєму складі таку саму кількість
індикатора та 1-2 краплі титранту (наприклад, при титруванні барбіталу в
спиртово-водному середовищі).
Наведені далі методи можна розглядати як окремі випадки кислотно-
основного титрування у водному середовищі.
3.2.4.1. Титрування кислот (алкаліметрія)
Титрування сильних кислот
Визначення кислоти хлороводневої. Хлороводнева кислота належить до
сильних кислот, тому підбір індикатору не викликає ускладнень: для цього
можна рекомендувати метиленовий оранжевий, перехід забарвлення якого
лежить в кислому середовищі:
HCl + NaOH → NaCl + H2O
Методика. В невелику конічну колбу з притертою пробкою наливають 10 мл
води й точно зважують, потім додають 3 мл розчину кислоти хлороводневої,
добре перемішують, закривають пробкою і знову точно зважують. Вміст колби
титрують 1 моль/л розчином натрію гідроксиду до переходу рожевого
забарвлення в оранжево-жовте (індикатор – метиловий оранжевий).
Е=М.м.; 1 мл 1 моль/л розчину натрію гідроксиду відповідає 0,03646 г
кислоти хлороводневої, якої в субстанції повинно бути не менш 24,8% і не
більш 25,2%.
Розрахунок кількісного вмісту HCl у відсотках проводять за
формулою 3.5.
Титрування слабких кислот
Визначення кислоти нікотинової. Оскільки водні розчини солей слабких
кислот внаслідок гідролізу мають лужну реакцію середовища, для титрування
слабких кислот більш придатними є індикатори з переходом забарвлення в
лужному середовищі (індикатор – фенолфталеїн):
Методика. Близько 0,3 г субстанції (точна наважка) вміщують в конічну колбу
ємністю 100 мл, розчиняють у 25 мл свіжопрокип’яченої гарячої води і по
охолодженні титрують 0,1 моль/л розчином натрію гідроксиду до рожевого
забарвлення, що не зникає протягом 1-2 хвилин (індикатор – фенолфталеїн).
Е=М.м.; 1 мл 0,1 моль/л розчину натрію гідроксиду відповідає 0,01231 г
кислоти нікотинової, кількісний вміст якої в субстанції має бути не менш 99,5%
у перерахунку на суху речовину.
Розрахунок кількісного вмісту кислоти нікотинової у відсотках проводять
за формулою 3.9.
+ H2O+ NaOH
N N
COOH COONa
87.
88
Визначення кислоти бензойної.Оскільки кислота бензойна у воді не
розчиняється, наважку розчиняють у спирті. До того ж в спиртовому
середовищі знижується ступінь гідролізу утвореної солі:
Методика. Близько 0,2 г субстанції (точна наважка) вміщують у 20 мл
нейтралізованого за фенолфталеїном спирту і титрують з тим же індикатором
0,1 моль/л розчином натрію гідроксиду до рожевого забарвлення.
Е=М.м.; 1 мл 0,1 моль/л розчину натрію гідроксиду відповідає 0,01221 г
кислоти бензойної, кількісний вміст якої в субстанції має бути не менш 99,5%.
Розрахунок кількісного вмісту кислоти бензойної у відсотках проводять
за формулою 3.5.
Визначення барбіталу. Барбітал є дуже слабкою кислотою (Ка=1,3.10-8
), тому
сіль, яка утворюється при його титруванні гідроксидом натрію, сильно
гідролізується, що призводить до появи лужної реакції розчину раніше
досягнення точки еквівалентності:
Використання спирту як розчинника для барбіталу, який не розчиняється
у воді, дозволяє також деякою мірою пригнітити гідроліз.
Як індикатор використовують тимолфталеїн, перехід забарвлення якого
(9,3-10,5) найбільш відповідає зміні pH в точці еквівалентності.
Методика. В дві однакові колби ємністю 100 мл вміщують по 10 мл спирту,
підлуженого за тимолфталеїном 0,1 моль/л розчином натрію гідроксиду до
стійкого блакитного забарвлення. В одну з колб вносять близько 0,5 г
субстанції (точна наважка), в іншу колбу додають 20 мл свіжопрокип’яченої та
охолодженої води. Одержаний розчин субстанції титрують 0,1 моль/л розчином
натрію гідроксиду до одержання забарвлення, однакового з забарвленням
контрольного розчину.
Е=М.м.; 1 мл 0,1 моль/л натрію гідроксиду відповідає 0,01842 г барбіталу,
кількісний вміст якого в субстанції має бути не менш 99,5%.
Розрахунок кількісного вмісту барбіталу у відсотках проводять за
формулою 3.5.
Визначення теофіліну. Дуже слабкі кислотні властивості теофіліну
(Ка=1,7.10-9
) не дають можливості безпосередньо відтитрувати його лугом,
тому використовують непряме титрування (за замісником). При взаємодії з
аргентуму нітратом утворюється комплексна сіль теофіліну і виділяється
+ H2O+ NaOH
COOH COONa
+ NaOH
N
N
H
O
O
C2H5
C2H5
O
H
+ H2O
N
N
NaO
H
O
O
C2H5
C2H5
88.
89
еквівалентна кількість нітратноїкислоти, яку й відтитровують натрію
гідроксидом:
Методика. Близько 0,4 г попередньо висушеної речовини (точна наважка)
розчиняють у 100 мл окропу (попередньо перевареного протягом 15 хвилин).
До охолодженого розчину додають 25 мл 0,1 моль/л розчину аргентуму нітрату,
1-1,5 мл розчину фенолового червоного й титрують 0,1 моль/л розчином натрію
гідроксиду до появи фіолетово-червоного забарвлення.
Е=М.м.; 1 мл 0,1 моль/л розчину натрію гідроксиду відповідає 0,01802 г
теофіліну, якого у висушеній субстанції повинно бути не менш 99,0%.
Розрахунок кількісного вмісту теофіліну у відсотках проводять за
формулою 3.5.
Визначення фенілсаліцилату. Карбоксильна група в молекулі
фенілсаліцилату заблокована залишком фенолу, тому речовину попередньо
омилюють точно виміряним об’ємом 0,5 моль/л розчину натрію гідроксиду.
Надлишок натрію гідроксиду відтитровують 0,5 моль/л розчином кислоти
хлороводневої. Індикатор – бромкрезоловий пурпуровий (pH=5,5-6,8), який
дозволяє відтитровувати не тільки надлишковий луг, але й луг, що зв’язаний з
фенольними гідроксилами, не титруючи луг, зв’язаний з карбоксильною
групою:
Методика. Близько 1 г субстанції (точна наважка) вміщують в конічну колбу
ємністю 100 мл, додають 25 мл 0,5 моль/л розчину натрію гідроксиду,
приєднують зворотній холодильник і, зануривши колбу в киплячий водяний
обігрівач, нагрівають до зникнення маслянистих крапель (1-1,5 год.). Розчин
охолоджують і надлишок лугу відтитровують 0,5 моль/л розчином кислоти
HNO3 + NaOH NaNO3 + H2O
+ HNO3
N
AgNN
N
O
O
H3C
CH3
+ AgNO3
N
NHN
N
O
O
H3C
CH3
NaCl ++ HCl
NaCl ++ HCl
NaOH + HCl NaCl + H2O
+ 2H2O++ 3NaOH
OH
COOC6H5
ONa
COONa ONa
ONa
COONa
OH
COONa
ONa OH
89.
90
хлороводневої до стійкогожовтого забарвлення (індикатор – бромкрезоловий
пурпуровий).
Паралельно проводять контрольний дослід.
Е=М.м.; 1 мл 0,5 моль/л розчину натрію гідроксиду відповідає 0,1071 г
фенілсаліцилату, кількісний вміст якого в субстанції має бути не менш 99,0%.
Розрахунок кількісного вмісту фенілсаліцилату у відсотках проводять за
формулою 3.8.
Титрування солей, утворених сильними кислотами та слабкими основами
До цієї категорії належать солі, які часто зустрічаються серед лікарських
речовин, утворені сильними мінеральними кислотами та органічними основами
(хоча слід відзначити, що серед органічних основ зустрічаються досить сильні).
Такі солі можна визначати алкаліметрично. Якщо сіль утворена дуже слабкою
основою (папаверину гідрохлорид, дибазол) то титрування проводять у водно-
спиртовому середовищі (спирт сприяє зниженню основності органічної основи,
яка утворюється в процесі титрування). Якщо ж сіль утворено основою, сила
якої достатня, щоб змінити забарвлення індикатору (атропіну сульфат, хініну
гідрохлорид та ін.), то титрування проводять у присутності не лише спирту, а й
органічного розчинника, який не змішується з водою (хлороформ, ефір та ін.).
Основа, що виділилась, екстрагується органічним розчинником, і таким чином,
не впливає на індикатор.
Визначення атропіну сульфату. Атропіну сульфат утворено досить сильною
основою (Кв=4,35), тому титрування проводять у присутності спирто-
хлороформної суміші:
Методика. Близько 0,5 г субстанції (точна наважка) розчиняють у 10 мл
спирту, нейтралізованого за фенолфталеїном, додають 20 мл хлороформу й
титрують 0,1 моль/л розчином натрію гідроксиду при сильному струшуванні до
появи блідо-рожевого забарвлення водного шару (індикатор – фенолфталеїн).
Е=1/2М.м.; 1 мл 0,1 моль/л розчину натрію гідроксиду відповідає
0,03384 г безводного атропіну сульфату, якого у перерахунку на суху речовину
повинно бути не менш 99,3%.
Розрахунок кількісного вмісту атропіну сульфату у відсотках проводять
за формулою:
)100(
100100
%
Bm
TKV
X
−⋅
⋅⋅⋅⋅
= , (3.9)
N CH3 O C
O
CH
CH2OH
C6H5
2
H2SO4
. + 2NaOH 2 C6H5CH
CH2OH
C
O
OCH3N +
+ Na2SO4 + 2H2O
90.
91
де V –об’єм 0,1 моль/л розчину натрію гідроксиду, витрачений на
титрування, мл;
К – коефіцієнт поправки 0,1 моль/л розчину натрію гідроксиду;
Т – титр 0,1 моль/л розчину натрію гідроксиду за атропіну
сульфатом, г/мл;
m – маса наважки атропіну сульфату, г;
B – процент вологості атропіну сульфату.
3.2.4.2. Титрування основ (ацидиметрія)
Ацидиметричні методи титрування у фарманалізі використовують для
визначення органічних основ або солей, утворених сильними основами та
слабкими кислотами. Титрантами є водні розчини сильних кислот –
хлороводневої та сульфатної. Для визначення слабких основ зазвичай
підбирають індикатор з переходом забарвлення в кислотному середовищі:
метиловий оранжевий, метиловий червоний та ін.
Титрування основ
Визначення кодеїну. Титрування відбувається за реакцією:
Методика. Близько 0,3 г субстанції (точна наважка) розчиняють при слабкому
нагріванні в 2 мл спирту, нейтралізованого за метиловим червоним, додають
20 мл тільки-но перевареної води та титрують 0,1 моль/л розчином
хлороводневої кислоти до рожевого забарвлення (індикатор – метиловий
червоний).
Е=М.м.; 1 мл 0,1 моль/л розчину кислоти хлороводневої відповідає
0,02994 г кодеїну, якого у перерахунку на суху речовину повинно бути не менш
99,0%.
Розрахунок кількісного вмісту кодеїну у відсотках проводять за
формулою 3.9.
Визначення гексаметилентетраміну. Речовину гідролізують при кип’ятінні з
сульфатною кислотою, після чого надлишок кислоти відтитровують лугом:
надл. H2SO4 + 2NaOH Na2SO4 + 2H2O
tº
(CH2)6N4 + 2H2SO4 + 6H2O 6CH2O + 2(NH4)2SO4
HCl+
H3CO
O
H
HO
N CH3N
HO
O
H3CO
CH3
+
Cl
-
91.
92
Методика. Близько 0,12г субстанції (точна наважка) розчиняють в конічній
колбі в 10 мл води, додають 50 мл 0,1 моль/л сульфатної кислоти; суміш
кип’ятять на невеликому вогні протягом 30 хвилин та охолоджують. До
охолодженої рідини додають 2 краплі розчину метилового червоного і
надлишок сульфатної кислоти відтитровують 0,1 моль/л розчином натрію
гідроксиду до жовтого забарвлення.
Паралельно проводять контрольний дослід.
Е=1/4М.м.; 1 мл 0,1 моль/л розчину сульфатної кислоти відповідає
0,003505 г гексаметилентетраміну, кількісний вміст якого має бути не менш
99,0%.
Розрахунок кількісного вмісту гексаметилентетраміну у відсотках
проводять за формулою 3.8.
Титрування солей, утворених сильною основою та слабкою кислотою
Солі, утворені сильними основами та слабкими кислотами у водних
розчинах гідролізуються з утворенням лужного середовища, що дозволяє
відтитровувати їх кислотою.
Визначення барбітал-натрію. Титрування відбувається за реакцією:
Методика. Близько 0,5 г субстанції (точна наважка) розчиняють у 30 мл
тільки-но перевареної та охолодженої води і титрують 0,1 моль/л розчином
кислоти хлороводневої до виникнення рожевого забарвлення (індикатор –
метиловий оранжевий).
Е=М.м.; 1 мл 0,1 моль/л розчину кислоти хлороводневої відповідає
0,02062 г барбітал-натрію, кількісний вміст якого повинен бути не менш 98,5%.
Розрахунок кількісного вмісту барбітал-натрію у відсотках проводять за
формулою 3.5.
При наявності в субстанції вільного лугу від знайденого процентного
вмісту барбітал-натрію віднімають вміст вільного лугу, помножений на
коефіцієнт 5,15 (відношення молекулярної маси барбітал-натрію до
молекулярної маси натрію гідроксиду).
Визначення натрію бензоату. Оскільки бензойна кислота, що виділяється в
процесі реакції здатна вплинути на індикатор, титрування ведуть у присутності
ефіру, який екстрагує утворену кислоту:
N C
CNH
O
C2H5
C2H5
O
HO + NaCl+ HCl
N C
CNH
NaO
O
C2H5
C2H5
O
NaOH + HCl H2O + NaCl
+ NaOH
N C
CNH
O
C2H5
C2H5
O
HO+ H2O
N C
CNH
NaO
O
C2H5
C2H5
O
Сумарно:
92.
93
Методика. Близько 1,5г субстанції (точна наважка) розчиняють у 20 мл води в
колбі з притертою пробкою ємністю 250 мл, додають 45 мл ефіру, 3-4 краплі
змішаного індикатору (1 мл розчину метилового оранжевого і 1 мл розчину
метиленового синього) і титрують 0,5 моль/л розчином кислоти хлороводневої
до появи бузкового забарвлення водного шару. В кінці титрування вміст колби
добре перемішують.
Е=М.м.; 1 мл 0,5 моль/л розчину кислоти хлороводневої відповідає
0,07205 г натрію бензоату, якого в перерахунку на суху речовину повинно бути
не менш 99,0%.
Розрахунок кількісного вмісту натрію бензоату у відсотках проводять за
формулою 3.9.
Непряме ацидиметричне титрування (за замісником)
Визначення меркурію окису жовтого. При взаємодії меркурію(ІІ) оксиду з
розчином калію йодиду утворюється розчинна комплексна сіль та луг, який
відтитровують кислотою:
HgО + 4KI + H2O → K2[HgI4] + 2KOH
2KOH + 2HCl → 2KCl + 2H2O
Сумарно: HgO + 4KI + 2HCl → K2[HgI4] + 2KCl + H2O
Методика. Близько 0,2 г субстанції (точна наважка) збовтують з 5 мл води,
додають 2 г калію йодиду та знову збовтують до повного розчинення. Після
цього додають ще 20 мл води і титрують 0,1 моль/л розчином кислоти
хлороводневої до появи рожевого забарвлення (індикатор – метиловий
червоний).
Е=1/2М.м.; 1 мл 0,1 моль/л розчину кислоти хлороводневої відповідає
0,01083 г меркурію окису, кількісний вміст якого повинен бути не менш 99,0%.
Розрахунок кількісного вмісту меркурію окису жовтого ведуть за
формулою 3.5.
3.2.5. Кислотно-основне титрування в неводному середовищі
Метод кислотно-основного титрування в неводному середовищі
застосовується для кількісного визначення лікарських речовин, які є слабкими
основами або кислотами (Кдис.<1.10-8
), їх солей, а також речовин, які погано
розчиняються у воді.
Під впливом різних розчинників властивості однієї і тієї ж речовини
можуть різко змінюватися. Сила кислоти або основи визначається ступенем їх
взаємодії з розчинником. Правильно підібраний неводний розчинник може
посилювати основні або кислотні властивості слабкої основи або слабкої
+ NaCl+ HCl
COONa COOH
93.
94
кислоти, що робитьможливим їх кількісне визначення кислотно-основним
титруванням.
За характером участі в кислотно-основному процесі всі розчинники
поділяють на дві великі групи: апротонні та протеолітичні.
Апротонні розчинники – це хімічні сполуки, молекули яких не іонізовані
й не здатні ні віддавати, ні приєднувати протон. Вони не вступають у взаємодію
з розчиненою речовиною (бензол, толуол, гексан, дихлоретан, хлороформ,
чотирихлористий вуглець). Апротонні розчинники часто додають до
іонізуючих розчинників для пригнічення сольволізу (термін, який відповідає
гідролізу у водному середовищі), що сприяє більш чіткому встановленню кінця
титрування.
Протолітичні розчинники – це хімічні сполуки здатні віддавати або
приєднувати протони. Їх в свою чергу поділяють на три групи:
1. Амфіпротні розчинники, які можуть як віддавати, так і приєднувати протон
(вода, одно- та багатоатомні спирти, інші сполуки). Амфіпротні розчинники
використовують для титрування речовин як кислотного, так і основного
характеру.
2. Протогенні або кислі розчинники, у яких здатність віддавати протон значно
перевищує здатність його приєднувати (мурашина, оцтова, пропіонова та
інші кислоти). Протогенні розчинники посилюють основні властивості
сполук.
3. Протофільні або основні розчинники, у яких акцепторні властивості по
відношенню до протона переважають над донорними (піридин,
диметилформамід, етилендіамін, діоксан і т. ін).
Критерієм можливості проведення кислотно-основного титрування та
правильності вибору розчинника служить константа титрування КТ (окремий
випадок константи рівноваги), яка визначається двома основними величинами:
– константою дисоціації розчиненої речовини (Ка або Кb);
– константою автопротолізу розчинника або іонним добутком розчинника (Кі).
Для спрощення розрахунків застосовуються величини від’ємних
логарифмів цих констант – рКТ, рКа, рКb, рКі.
В довідниках наводяться значення констант дисоціації кислот і основ в
різних неводних розчинниках і константи іонних добутків різних неводних
розчинників. Використовуючи їх, можна розрахувати в кожному окремому
випадку рКТ:
– для кислот: КТ = Кі / Ка або рКТ= рКі – рКа
– для основ: КТ = Ка або рКТ = рКа
Чим більшою є величина рКТ, тим кращі умови титрування.
Найкращі умови титрування слабких кислот досягаються в основних
неводних розчинах, таких як піридин, диметилформамід; слабких основ – в
кислих неводних розчинниках, таких як оцтова кислота, оцтовий ангідрид.
Солі органічних та деяких мінеральних кислот можуть бути визначені також, як
і основи титруванням у кислих розчинниках.
94.
95
При титруванні сумішікислот або суміші основ застосовують
диференціюючі розчинники з величиною рКі, що перевищує 15, які не мають
виражених кислотно-основних властивостей (табл. 3.2).
Кінцеву точку титрування визначають за допомогою індикаторів (див.
табл. 3.2) або потенціометрично.
Таблиця 3.2
Розчинники Індикатори Титранти
Кислі
Оцтова та мурашина кис-
лоти, оцтовий ангідрид та
їхні суміші з іншими
розчинниками
Кристалічний фіолетовий,
судан III, тропеолін 00, мети-
ловий фіолетовий, нейтраль-
ний червоний, малахітовий
зелений, диметиламіноазо-
бензол
Розчин хлорної кислоти в оцто-
вій кислоті або в нітрометані
Основні
Диметилформамід,
піридин, етилендіамін
Тимоловий синій, бромтимо-
ловий синій, α-нафтолбензеїн,
орто-нітроанілін
Розчини натрію гідроксиду,
калію гідроксиду, метилату
натрію, метилату літію,
гідроксиду тетраетиламонію в
метиловому спирті або в суміші
метилового спирту та бензолу.
Диференціюючі
Ацетон, діоксан, нітроме-
тан, метилетилкетон, ме-
тиловий спирт, ізопропі-
ловий спирт, третинний
бутиловий спирт, диме-
тилсульфоксид
Метиловий оранжевий, тимо-
ловий синій, бромфеноловий
синій, нейтральний червоний,
метиловий червоний,
бромтимоловий синій
Розчини хлороводневої кислоти
в метиловому спирті або в
гліколевих сумішах; розчини
хлорної кислоти в нітрометані,
в метиловому спирті або в
гліколевих сумішах; розчини,
які застосовуються при титру-
ванні в основних розчинниках
3.2.5.1. Титрування органічних основ та їх солей (ацидиметрія)
Титрування слабких органічних основ
Для поліпшення умов титрування лікарських речовин, які є слабкими
органічними основами, частіше як розчинник застосовують безводну (льодяну)
оцтову кислоту, оцтовий ангідрид або їх суміш. У деяких випадках додають
апротонні розчинники. Льодяна оцтова кислота здатна бути донором протонів і,
таким чином, збільшувати силу розчинених основ.
Оцтовий ангідрид підвищує кислотність та діелектричну проникність
середовища, апротонні розчинники знижують її іонний добуток.
Для фіксування кінцевої точки титрування застосовують частіше за все
індикатор кристалічний фіолетовий (розчин в льодяній оцтовій кислоті):
перехід забарвлення від фіолетового (лужне середовище) через синьо-зелене
(нейтральне) до зеленкувато-жовтого (кисле середовище) і метиловий
оранжевий (в ацетоні): перехід забарвлення від жовтого до рожевого.
Визначення кофеїну. Кофеїн дуже слабка основа (pK=13,40), тому для його
визначення з достатньою точністю титрування проводять в оцтовому ангідриді
з додаванням апротонного розчинника – бензолу, який дозволяє більш чітко
95.
96
зафіксувати кінцеву точкутитрування. Речовину попередньо висушують для
видалення води, яка може погіршити умови титрування. Кофеїн протонується
розчинником, при цьому оцтовий ангідрид перетворюється в аніон. Хлорна
кислота реагує з льодяною оцтовою кислотою, яка є її розчинником з
утворенням іону ацетонію. Потім аніон оцтового ангідриду реагує з іоном
ацетонію:
Е=М.м.
Методика. Близько 0,15 г попередньо висушеної при 80оС до сталої маси
субстанції (точна наважка) розчиняють у 10 мл оцтового ангідриду при
нагріванні на водяному обігрівачі, додають 20 мл бензолу, 5 крапель розчину
кристалічного фіолетового і титрують 0,1 моль/л розчином хлорної кислоти до
одержання жовтого забарвлення.
Паралельно проводять контрольний дослід.
1 мл 0,1 моль/л розчину хлорної кислоти відповідає 0,01942 г безводного
кофеїну, якого у висушеній субстанції повинно бути не менш 99,0%.
Розрахунок кількісного вмісту кофеїну у відсотках проводять за
формулою:
m
TKVV
X к 100)(
%
⋅⋅⋅−
= , (3.10)
де V – об’єм 0,1 моль/л розчину хлорної кислоти, витраченого на
титрування наважки, мл;
Vк – об’єм 0,1 моль/л розчину хлорної кислоти, витраченого на
титрування у контрольному досліді, мл;
К – коефіцієнт поправки до молярності 0,1 моль/л розчину хлорної
кислоти;
Т – титр 0,1 моль/л розчину хлорної кислоти за кофеїном, г/мл;
m – маса наважки висушеного кофеїну, г.
(CH3C
O
O CCH2) -
O
CCH2) -
O
+ (CH3C
O
O
ClO4
-
N
N
CH3
H
N
NO
O
CH3
H3C
+
+ ClO4
-
N
N
CH3
H
N
NO
O
CH3
H3C
+
+ CH3COOH2
+ (CH3CO)2O + CH3COOH
+
N
NO
O
CH3
H3C N
N
CH3
H
N
N
CH3N
NO
O
CH3
H3C
(CH3CO)2O +
CH3COOH + HClO4 CH3COOH2
+ + ClO4
-
96.
97
Титрування солей органічнихоснов
Солі слабких основ можна, як і основи, титрувати в середовищі льодяної
оцтової кислоти розчином хлорної кислоти.
При титруванні солей галоїдводневих кислот (гідрохлоридів,
гідробромідів, гідройодидів) титрування ведуть у присутності меркурію (ІІ)
ацетату, який зв’язує галоїд у малодисоційовану сполуку (HgHal2), запобігаючи
тим самим утворенню галоїдводнів, які навіть у середовищі безводної оцтової
кислоти мають ступінь дисоціації, достатній для того, щоб привести до зміни
забарвлення індикатора.
Сульфатна кислота по другому ступеню, нітратна, фосфорна та органічні
кислоти в середовищі льодяної оцтової кислоти практично не проявляють
кислотних властивостей, тому при визначенні їх солей не заважають
встановленню моменту еквівалентності.
Визначення промедолу. При титруванні промедолу, який є сіллю органічної
основи, відбуваються такі хімічні реакції:
Е=М.м.
Сумарна реакція:
. HCl2
N
OCOC6H5H5C6
CH3
CH3
H3C
+ 2HClO4 + Hg(CH3COO)2 2
+
N
H5C6
CH3
CH3
H3C
H
OCOC6H5
+
+ HgCl2 + 2CH3COOH
ClO4
+ 4CH3COOH
N
H5C6
CH3
CH3
H3C
H
OCOC2H5
+
2
N
OCOC6H5H5C6
CH3
CH3
H3C
H
+
2 CH3COO
-
+ HgCl2CH3COO
-
2
+
N
OCOC6H5H5C6
CH3
CH3
H3C
H
+ Hg(CH3COO)2
. HCl2
N
OCOC6H5H5C6
CH3
CH3
H3C
2HClO4 + 2CH3COOH 2CH3COOH2
+ + 2ClO4
-
+ 2CH3COOH2
+
ClO42
+
N
H5C6
CH3
CH3
H3C
H
OCOC2H5
+ 2ClO42 +
N
OCOC6H5H5C6
CH3
CH3
H3C
H
97.
98
Методика. Близько 0,3г субстанції (точна наважка) розчиняють у 20 мл
льодяної оцтової кислоти, додають 5 мл розчину меркурію (ІІ) ацетату і
титрують 0,1 моль/л розчином хлорної кислоти до появи зеленого забарвлення
(індикатор – кристалічний фіолетовий).
Паралельно проводять контрольний дослід.
1 мл 0,1 моль/л розчину хлорної кислоти відповідає 0,03118 г промедолу,
якого в субстанції повинно бути не менш 99,0% і не більш 101,%.
Розрахунок кількісного вмісту промедолу у відсотках проводять за
формулою 3.10.
Визначення атропіну сульфату. Сульфат-іон у середовищі льодяної оцтової
кислоти є однокислотною основою, він реагує з іоном ацетонію з утворенням
гідросульфат-іону. Гідросульфат-іон не проявляє основних властивостей по
відношенню до іону ацетонію:
Тому при титруванні атропіну сульфату утворюються еквівалентні
кількості атропіну перхлорату та атропіну гідросульфату:
Е=М.м.
SO4
2- + CH3COOH2
+ HSO4
-
+ CH3COOH
HSO4
-
+ CH3COOH2
+
+
N CH3
H
O C CH
O CH2OH
C6H5
+ CH3COO
-
+
+
O C CH
O CH2OH
C6H5N CH3
H
HClO4 + CH3COOH ClO4
-
+ CH3COOH2
+
CH3COO
-
+ CH3COOH2
+ 2CH3COOH
+
+O C CH
O CH2OH
C6H5N CH3
H
+ CH3COOH
2
N CH3
H
O C CH
O CH2OH
C6H5
+
N CH3
H
O C CH
O CH2OH
C6H5
+
+ ClO4
-
SO4
2-
HSO4
-
ClO4
-
N CH3
H
O C CH
O CH2OH
C6H5
2
+
O C CH
O CH2OH
C6H5N CH3
H
+
+
+
O C CH
O CH2OH
C6H5N CH3
H
+ HClO4
Сумарна реакція:
+ ClO4
-
HSO4
-
SO4
2-
98.
99
Методика. Близько 0,5г висушеної при 100-105о
С до сталої маси субстанції
(точна наважка) розчиняють у 10 мл безводної оцтової кислоти при слабкому
нагріванні на водяному обігрівачі. До охолодженого розчину додають 3 краплі
розчину кристалічного фіолетового і титрують 0,1 моль/л розчином хлорної
кислоти до появи зеленого забарвлення.
Паралельно проводять контрольний дослід.
1 мл 0,1 моль/л розчину хлорної кислоти відповідає 0,06768 г атропіну
сульфату, якого у висушеній субстанції повинно бути не менш 99,0%.
Розрахунок кількісного вмісту атропіну сульфату у відсотках проводять
за формулою 3.10.
Визначення аміназину. Іноді гідрохлориди органічних основ титрують в
середовищі оцтового ангідриду. В цьому випадку додавання меркурію (ІІ)
ацетату не потрібно:
Е=М.м.
ClO4
-
+
+ CH3COCl + CH3COOH
+ HClO4 + (CH3CO)2OCl
-
Сумарна реакція:
N
S
Cl
CH2 (CH2)2 N
CH3
CH3H
N
S
Cl
CH2 (CH2)2 N
CH3
CH3H
+
+
N
S
Cl
CH2 (CH2)2 N
CH3
CH3H
+
HClO4 + CH3COOH ClO4 + CH3COOH2
+Cl
-
+ (CH3CO)2O
+ CH3 C
O
CI
+ CH3COO
-
+ ClO4
-
CH3COO
-
+ CH3COOH2
+ 2CH2COOH
N
S
Cl
CH2 (CH2)2 N
CH3
CH3H
+
N
S
Cl
CH2 (CH2)2 N
CH3
CH3H
+
N
S
Cl
CH2 (CH2)2 N
CH3
CH3H
+
ClO4
-
99.
100
Методика. Близько 0,3г субстанції (точна наважка) розчиняють у 30 мл
оцтового ангідриду і титрують 0,1 моль/л розчином хлорної кислоти до
зеленого забарвлення (індикатор – кристалічний фіолетовий).
Паралельно проводять контрольний дослід.
1 мл 0,1 моль/л розчину хлорної кислоти відповідає 0,03553 г аміназину,
якого у перерахунку на суху речовину повинно бути не менш 99,0% і не більш
101,0%.
Розрахунок кількісного вмісту аміназину у відсотках проводять за
формулою:
)100(
100100)(
%
Bm
TKVV
X к
−⋅
⋅⋅⋅⋅−
= , (3.11)
де V – об’єм 0,1 моль/л розчину хлорної кислоти, витраченого на
титрування наважки, мл;
Vк – об’єм 0,1 моль/л розчину хлорної кислоти, витраченого на
титрування у контрольному досліді, мл;
К – коефіцієнт поправки до молярності 0,1 моль/л розчину хлорної
кислоти;
Т – титр 0,1 моль/л розчину хлорної кислоти за аміназином, г/мл;
m – маса наважки аміназину, г;
В – процент вологості аміназину.
3.2.5.2. Титрування кислот (алкаліметрія)
Для визначення слабких кислот, які неможливо відтитрувати у воді
(карбонові кислоти, амінокислоти, барбітурати, теобромін, теофілін та ін.),
застосовують розчинники основного характеру – ДМФА, піридин,
етилендіамін. Ці розчинники, маючи протоноакцепторні властивості,
приєднують протон слабкої кислоти, тим самим посилюючи її кислотні
властивості. Як титрант застосовують титровані розчини метилату натрію,
розчини натрію або тетраетиламонію гідроксиду в суміші бензолу та метанолу.
При титруванні в основних розчинниках слід захищати розчин, що
титрується, а також титрант від двоокису вуглецю, який є у повітрі. Титрування
проводять у закритому посуді, іноді в атмосфері інертного газу.
Індикатор – частіше за все тимоловий синій (перехід забарвлення від
жовтого до синього).
Визначення барбіталу. Титрування відбувається за реакцією:
C
CNH
O
-
O
C2H5
C2H5
O
N
("онієвий" іон)
сольватований протон
+
+ HC N
H
O CH3
CH3
+ H C
O
N
CH3
CH3NH C
CNH
C
O
C2H5
C2H5
O
O
100.
101
Е=М.м.
Методика. Близько 0,15г субстанції (точна наважка) розчиняють в 10 мл
суміші диметилформаміду і бензолу (1:3) попередньо нейтралізованій за
тимоловим синім у диметилформаміді, та титрують з тим же індикатором з
напівмікробюретки 0,1 моль/л розчином натрію гідроксиду в суміші
метилового спирту та бензолу до синього забарвлення.
1 мл 0,1 моль/л розчину натрію гідроксиду відповідає 0,01842 г барбіталу,
якого в субстанції повинно бути не менш 99,0% і не більш 101,0%.
Розрахунок кількісного вмісту барбіталу у відсотках проводять за
формулою 3.5.
3.2.6. Методи окиснення-відновлення
Ці методи базуються на застосуванні окисно-відновних реакцій, тобто
реакцій, пов’язаних з переносом електронів.
Вони є дуже розповсюдженими методами титриметричного аналізу і
дозволяють прямо або зворотно визначати практично всі неорганічні лікарські
речовини, які здатні, за певних умов, стехіометрично приймати або віддавати
електрони, тобто бути окисниками або відновниками. Крім того, методи
окисно-відновного титрування придатні для визначення багатьох органічних
лікарських речовин, які є потенційними відновниками і тому можуть бути
окиснені до речовин з меншою відновною здатністю, ніж вихідні речовини.
Кінцеву точку титрування в окисно-відновних методах визначають за
допомогою редокс-індикаторів – речовин, здатних в середовищі з певним
окисно-відновним потенціалом окиснюватись і змінювати своє забарвлення, а
також специфічних індикаторів (наприклад, метиловий червоний у
броматометрії; крохмаль в йодометрії).
Значення молярної маси еквіваленту для лікарської речовини в цих
методах знаходять шляхом ділення її молекулярної маси на число електронів,
які приймає або віддає речовина у відповідній хімічній реакції.
В фармацевтичному аналізі найчастіше застосовуються
перманганатометрія, йодометрія, броматометрія, нітритометрія та інші.
3.2.6.1. Перманганатометрія
Метод базується на застосуванні реакції окиснення лікарської речовини,
що визначається, перманганат-іонами. Найбільш часто в титриметричному
аналізі застосовують реакції окиснення перманганат-іонами в сильно кислому
середовищі. Концентрація кислоти повинна бути не менше 1 моль/л. Це
зумовлено тим, що величина редокс-потенціалу системи MnО4
-/Mn2+
дуже
сильно залежить від концентрації кислоти.
+ H2O+ HC N
O CH3
CH3C
CNH
O
C2H5
C2H5
O
N
NaO+ HC N
H
O CH3
CH3
+
C
CNH
O
-
O
C2H5
C2H5
O
N
+ NaOH
101.
102
Для створення кислогосередовища застосовують сульфатну кислоту.
Хлороводневу кислоту не застосовують, оскільки хлорид-іони проявляють
відновні властивості й можуть бути окиснені перманганат-іонами до хлору .
Нітратна кислота сама є окисником і може викликати побічні реакції,
тому її теж не застосовують.
Основним рівнянням перманганатометрії є:
Розчин калію перманганату інтенсивно забарвлений у червоно-
фіолетовий колір. Навіть одна крапля 0,01 моль/л розчину забарвлює розчин,
що титрується, в помітно рожевий колір, тому спеціальних індикаторів у
перманганатометрії не застосовують.
Нормальний окисно-відновний потенціал MnО4
-/Mn2+
становить 1,51 В, у
зв’язку з чим розчини калію перманганату в кислому середовищі можна
застосовувати для визначення лікарських речовин, які не взаємодіють з більш
слабкими окисниками.
Методом перманганатометрії визначають кількісний вміст розчину
перекису водню, магнію перекису, натрію нітриту.
Визначення розчину перекису водню. В основі цього методу лежить реакція:
5H2O2 + 2KMnO4 + 3H2SO4 → 2MnSO4 + K2SO4 + 5O2↑ + 8H2O
Методика. Точно відмірюють 10 мл 3% розчину перекису водню, вміщують в
мірну колбу ємністю 100 мл і доводять об’єм розчину водою до мітки. До 10 мл
одержаного розчину додають 5 мл розведеної сульфатної кислоти і титрують
0,1 моль/л розчином калію перманганату до блідо-рожевого забарвлення.
Е=1/2М.м.; 1 мл 0,1 моль/л розчину калію перманганату відповідає
0,001701 г перекису водню, якого в субстанції повинно бути не менш 2,7-3,3 %.
Розрахунок кількісного вмісту перекису водню у відсотках проводять за
формулою:
,
100
%
1 п
МК
VV
VTKV
X
⋅
⋅⋅⋅⋅
= (3.12)
де V – об’єм 0,1 моль/л розчину калію перманганату, мл;
Vмк – об’єм мірної колби, мл;
V1 – об’єм розчину перекису водню, взятого для аналізу, мл;
Vп – об’єм піпетки, мл;
К – коефіцієнт поправки до молярності 0,1 моль/л розчину
калію перманганату;
Т – титр 0,1 моль/л розчину калію перманганату за перекисом водню,
г/мл.
MnO4
-
+ 5e + 8H
+
Mn2+ + 4H2O
102.
103
Визначення натрію нітриту.Натрію нітрит в кислому середовищі
розкладається з виділенням нітрозних газів, тому метод прямого титрування
застосовувати не можна. В кількісному визначенні натрію нітриту
використовують метод зворотного титрування з йодометричним визначенням
надлишку калію перманганату:
2KMnO4 + 5NaNO2 + 3H2SO4 → K2SO4 + 2MnSO4 + 5NaNO3 + 3H2O
2KMnO4 + 10KI + 8H2SO4 → 6K2SO4 + 5I2 + 2MnSO4 + 8H2O
I2 + 2Na2S2O3 → 2NaI + Na2S4O6
Методика. Близько 1 г субстанції (точна наважка) розчиняють в мірній колбі
ємністю 100 мл і розчин доводять водою до мітки. Точно відмірюють 10 мл
цього розчину і повільно виливають в суміш з 40 мл 0,1 моль/л розчину калію
перманганату, 300 мл води та 25 мл розведеної сульфатної кислоти. Через 20 хв
до одержаної рідини додають 2 г калію йодиду і йод, що виділився, титрують
0,1 моль/л розчином натрію тіосульфату (індикатор – крохмаль).
Паралельно проводять контрольний дослід.
Е=1/2М.м.; 1 мл 0,1 моль/л розчину натрію тіосульфату відповідає
0,003450 г натрію нітриту, якого в субстанції повинно бути не менш 98%.
Розрахунок кількісного вмісту натрію нітриту у відсотках проводять за
формулою:
п
мкк
Vm
VTKVV
X
⋅
⋅⋅⋅⋅−
=
100)(
% , (3.13)
де Vк – об’єм 0,1 моль/л розчину натрію тіосульфату, витраченого на
титрування у контрольному досліді, мл;
V – об’єм 0,1 моль/л розчину натрію тіосульфату, витраченого на
титрування в прямому досліді, мл;
Vмк – об’єм мірної колби, мл;
Vп – об’єм піпетки, мл;
К –коефіцієнт поправки до молярності 0,1 моль/л розчину
натрію тіосульфату;
Т – титр 0,1 моль/л розчину натрію тіосульфату за натрію нітритом, г/мл;
m – маса наважки натрію нітриту, г.
3.2.6.2. Йодометрія
Йодометрія – метод кількісного визначення вільного йоду, тих речовин,
які кількісно виділяють його під час реакцій і тих сполук, які зв’язують йод або
окиснюються йодом в стехіометричних кількостях.
Йодометричний метод кількісного визначення має широке практичне
застосування; за своєю простотою і точністю він визнається одним з кращих
редокс-методів кількісного визначення.
В основі йодометричного визначення лежать реакції:
[I3] + 2e 3I
3I
_
2e [I3]
103.
104
Нормальний окисно-відновний потенціалцієї системи дорівнює 0,545 В.
Ті речовини, які мають більш низький потенціал окиснюються йодом, а
речовини, що мають більш високий потенціал, окиснюють йодид-іони до йоду,
котрий потім може бути відтитрований за реакцією:
2S2O3
2-
+ [I3]- → S4O6
2-
+ 3I-
Нормальний окисно-відновний потенціал системи S4O6
2-
/2S2O3
2-
дорівнює
0,17 В.
Пряме йодометричне титрування. Методом прямого йодометричного
титрування визначають речовини, які мають сильні відновні властивості
(натрію тіосульфат, аскорбінова кислота, лікарські сполуки арсену (III) та інші).
Визначення проводять в кислому, нейтральному або слабко-лужному
середовищі. Титрантом є розчин йоду в йодиді калію. Цей розчин має жовто-
бурий колір і зайва його крапля забарвлює розчин, що титрується, в блідо-
жовтий колір, що може слугувати ознакою кінця титрування (кількісне
визначення анальгіну). Іноді рекомендують додавати декілька мілілітрів
органічного розчинника, що не змішується з водою, наприклад, хлороформу.
При збовтуванні надлишковий йод переходить в хлороформний шар і надає
йому фіолетового забарвлення.
Однак найбільш чітко кінцеву точку титрування можна визначити за
допомогою крохмалю, який з йодом в присутності йодид-іонів утворює
комплексну сполуку інтенсивно-синього кольору.
Зворотна йодометрія. Методом зворотної йодометрії визначають сполуки, які
повільно окиснюються йодом (ізоніазид), які утворюють з ним комплексні
сполуки (кофеїн), дають реакцію ароматичного заміщення (антипірин) або
потребують для стехіометричного необоротного окиснення лужного
середовища (формальдегід, глюкоза, фурацилін). В останньому випадку
окиснення відбувається за схемою:
Після завершення реакцій надлишок йоду відтитровують тіосульфатом
натрію. Якщо окиснення проводили в лужному середовищі, до реакційної
суміші спочатку додають надлишок кислоти, а тоді йод, що виділився,
відтитровують тіосульфатом натрію:
При проведенні зворотного йодометричного визначення крохмаль
додають в кінці титрування, коли розчин набуде блідо-жовтого кольору –
з’являється інтенсивне синє забарвлення – і далі титрують до знебарвлення.
Додавати крохмаль до розчинів з великою концентрацією йоду не можна,
оскільки в цьому випадку відбувається необоротне зв’язування йоду.
[I3] + 2OH 2I + IO + H2O
IO + H2O + 2e I + 2OH
[I3]IO + 2I + 2H+ + H2O
104.
105
Визначення окисників. Привизначенні речовин, які мають окиснювальні
властивості (калію перманганат, калію арсенат), до розчину речовини, як
правило в кислому середовищі, додають надлишок розчину йодиду калію. В
результаті окисно-відновної реакції виділяється еквівалентна кількість йоду,
який відтитровують розчином натрію тіосульфату. Індикатор – крохмаль.
Йодометричний метод застосовується також для визначення йодвмісних
органічних сполук після переведення йоду в іоногенний стан окисненням до
йодату (тиреойодин).
Визначення йоду. В основі визначення лежить реакція:
I2 + 2Na2S2O3 → 2NaI + Na2S4O6
Методика. В точно зважену колбу з притертою пробкою, яка містить 10 мл
розчину йодиду калію, висипають близько 0,2 г розтертого йоду і знову
зважують. Одержаний розчин розводять водою до 20 мл і титрують 0,1 моль/л
розчином натрію тіосульфату до знебарвлення (індикатор – крохмаль).
Е=1/2М.м.; 1 мл 0,1 моль/л розчину натрію тіосульфату відповідає
0,01269 г йоду, якого в субстанції повинно бути не менш 99,5%.
Розрахунок кількісного вмісту йоду у відсотках проводять за
формулою 3.5.
Визначення натрію тіосульфату. В основі визначення лежить реакція
взаємодії йоду з тіосульфатом натрію.
Методика. Близько 0,5 г речовини (точна наважка) розчиняють у 25 мл води і
титрують 0,1 моль/л розчином йоду (індикатор – крохмаль).
Е=М.м.; 1 мл 0,1 моль/л розчину йоду відповідає 0,02482 г натрію
тіосульфату, якого в субстанції повинно бути не менш 99,0% і не більш 102%.
Розрахунок кількісного вмісту натрію тіосульфату у відсотках проводять
за формулою 3.5.
Визначення розчину формальдегіду (аналогічно визначають глюкозу та інші
речовини, що містять альдегідну групу). В основі визначення лежить реакція
окиснення формальдегіду надлишком йоду:
Реакція оборотна. Щоб запобігти протіканню зворотної реакції
визначення необхідно проводити в лужному середовищі (для зв’язування
йодоводневої кислоти). Але луг взаємодіє також і з розчином йоду:
I2 + 2NaOH → NaI + NaOI + H2O
Гіпойодид натрію окиснює формальдегід:
C
O
H
H + I2 + H2O HCOOH + 2HI
C
O
H
H HCOONa + NaI + H2O+ NaIO + NaOH
105.
106
Після завершення процесуокиснення додають кислоту. Йод, який виділяється,
відтитровують тіосульфатом натрію:
NaIO + NaI + H2SO4 → I2 + Na2SO4 + H2O
I2 + 2Na2S2O3 → 2NaI + Na2S4O6
Методика. Близько 1 г субстанції (точна наважка) вміщують в мірну колбу
ємністю 100 мл і доводять водою до мітки. До 5 мл цього розчину в колбу з
притертою пробкою додають 20 мл 0,1 моль/л розчину йоду та 10 мл 1 моль/л
розчину натрію гідроксиду, збовтують і залишають у темному місці на
10 хвилин. Потім додають 11 мл 1 моль/л розчину сульфатної кислоти. Йод, що
виділився, титрують 0,1 моль/л розчином натрію тіосульфату до знебарвлення
(індикатор – крохмаль).
Е=1/2М.м.; 1 мл 0,1 моль/л розчину йоду відповідає 0,001501 г
формальдегіду, якого в субстанції повинно бути 36,5-37,5%.
Розрахунок кількісного вмісту формальдегіду у відсотках проводять за
формулою:
п
мк
Vm
VTKVKV
X
⋅
⋅⋅⋅⋅−⋅
=
100)(
% 2211
, (3.14)
де V1 – об’єм 0,1 моль/л розчину йоду, мл;
V2 – об’єм 0,1 моль/л розчину натрію тіосульфату, витраченого на
титрування надлишку йоду, мл;
Vмк – об’єм мірної колби, мл;
Vп – об’єм піпетки, мл;
К1 – коефіцієнт поправки до молярності 0,1 моль/л розчину йоду;
К2 – коефіцієнт поправки до молярності 0,1 моль/л розчину натрію
тіосульфату;
Т – титр 0,1 моль/л розчину йоду за формальдегідом, г/мл;
m – маса наважки формальдегіду, г.
3.2.6.3. Броматометрія
Пряме броматометричне титрування. Метод базується на застосуванні
окиснювальних властивостей бромат-іонів, які у кислому середовищі
відновлюються до бромід-іонів за таким рівнянням:
Ео
= 1,45 В
Титрування розчином KBrO3 виконують завжди у присутності KBr, при
цьому відбувається виділення вільного брому за рівнянням:
BrO3
- + 5Br- + 6H+
→ 3Br2 + 3H2O
Бром, який виділився, вступає в реакцію електрофільного заміщення або
виступає в ролі окисника:
Ео
= 1,065 В
6e + BrO3
-
+ 6H+ Br
-
+ 3H2O
Br2 + 2e 2Br
106.
107
Таким чином, підкисленірозчини KBrO3 та KBr діють як еквівалентні їм
розчини вільного брому. Фактично, вони є стійкими замінниками нестійких
розчинів брому.
В момент еквівалентності бром, який виділяється при додаванні
надлишкової краплі розчину KBrO3, забарвлює розчин, що титрується, у
жовтий колір. Найбільш чітко кінцеву точку титрування можна визначити за
допомогою кислотно-основних індикаторів: метилового червоного, метилового
оранжевого та інших, які в момент еквівалентності необоротно окиснюються
надлишком окисника і знебарвлюються.
У тих випадках, коли реакція протікає повільно, допускається нагрівання
до 50-60о
С.
Методом прямої броматометрії визначають, наприклад, лікарські
речовини, які мають у своєму складі арсен (III).
Визначення миш’яковистого ангідриду. Визначення ґрунтується на
окисненні оксиду арсену (ІІІ) калію броматом у кислому середовищі до оксиду
арсену (V). В момент еквівалентності зайва крапля калію бромату з бромідом
калію утворює вільний бром, який знебарвлює індикатор – метиловий
червоний:
3As2O3 + 2KBrO3 → 3As2O5 + 2KBr
KBrO3 + 5KBr + 3H2SO4 → 3Br2 + 3K2SO4 + 3H2O
Методика. Близько 0,1 г субстанції (точна наважка) розчиняють в 2-3 мл
розчину натрію гідроксиду. До розчину послідовно додають 50 мл води та
10 мл концентрованої сульфатної кислоти. Потім нагрівають до кипіння,
додають 0,5 г калію броміду і титрують 0,1 моль/л розчином калію бромату. В
кінці титрування додають 3 краплі метилового червоного і титрують по краплі
до знебарвлення індикатора.
Паралельно проводять контрольний дослід.
Е=1/2М.м.; 1 мл 0,1 моль/л розчину калію бромату відповідає 0,004946 г
оксиду арсену (ІІІ), якого в субстанції повинно бути не менш 99,0%.
Розрахунок кількісного вмісту миш’яковистого ангідриду у відсотках
проводять за формулою 3.10.
Визначення тимолу. Для кількісного визначення тимолу використовують
метод прямого титрування, оскільки в цьому випадку заміщення водню на бром
протікає досить швидко:
KBrO3 + 5KBr + 3H2SO4 → 3Br2 + 3K2SO4 + 3H2O
Е=1/4М.м.
+ 2HBr+ 2Br2
CH3
CH3C CH3
H
OH
CH3C CH3
H
Br
CH3
Br
OH
107.
108
Методика. Близько 0,5г субстанції (точна наважка) розчиняють в 5 мл розчину
натрію гідроксиду в мірній колбі ємністю 100 мл і доводять об’єм розчину
водою до мітки. Точно відмірені 10 мл одержаного розчину переносять в колбу
з притертою пробкою, додають 0,5 г броміду калію, 40 мл розведеної
хлороводневої кислоти, 3 краплі розчину метилового оранжевого і при
сильному збовтуванні титрують 0,1 моль/л розчином калію бромату. В кінці
титрування додають ще 2 краплі розчину метилового оранжевого. Зникнення
рожевого забарвлення рідини вказує на кінець титрування.
1 мл 0,1 моль/л розчину калію бромату відповідає 0,003755 г тимолу,
якого в субстанції повинно бути не менш 99,0%.
Розрахунок кількісного вмісту тимолу у відсотках проводять за
формулою 3.7.
Зворотна броматометрія. Методом зворотної броматометрії визначають
лікарські речовини, які повільно реагують з бромом, наприклад, сполуки здатні
вступати в реакцію електрофільного заміщення – реакцію бромування (феноли,
ароматичні аміни).
До розчину речовини, що визначається, додають розчини калію броміду,
калію бромату і сульфатної або хлороводневої кислоти. Виділяється бром, який
вступає в реакцію електрофільного заміщення. Як правило, реакція протікає
повільно, тому реакційну суміш залишають на деякий час для її завершення.
Надлишок брому визначають йодометрично – додають калію йодид і йод, що
виділився, відтитровують натрію тіосульфатом.
Визначення фенолу. Визначення ґрунтується на бромуванні фенолу:
KBrO3 + 5KBr + 3H2SO4 → 3Br2 + 3K2SO4 + 3H2O
Br2 + 2KI → 2KBr + I2
I2 + 2Na2S2O3 → 2NaI + Na2S4O6
Е=1/6М.м.
Методика. Близько 0,5 г субстанції (точна наважка) розчиняють у воді,
переносять в мірну колбу ємністю 250 мл та доводять водою до мітки.
Відмірюють 25 мл цього розчину, переносять його в склянку для бромування
ємністю 250 мл, додають 50 мл 0,1 моль/л розчину бромату калію, 1 г броміду
калію та 10 мл 50% розчину сульфатної кислоти. Рідину в склянці ретельно
перемішують і залишають на 15 хвилин. Потім до суміші додають 20 мл
розчину йодиду калію, сильно збовтують, залишають на 10 хвилин в темному
місці, після чого йод, який виділився, титрують 0,1 моль/л розчином натрію
тіосульфату (індикатор – крохмаль).
Паралельно проводять контрольний дослід.
+ 3HBr
Br
+ 3Br2
OH
Br
OH
Br
108.
109
1 мл 0,1моль/л розчину калію бромату відповідає 0,001568 г фенолу,
якого в субстанції повинно бути не менш 98,0%.
Розрахунок кількісного вмісту фенолу у відсотках проводять за
формулою 3.13.
3.2.6.4. Йодатометрія
Розчини калію йодату в практиці фармацевтичного аналізу
застосовуються для окиснювального титрування таких лікарських речовин, як
фтивазид, кислота аскорбінова, апресин та ін.
Йодат-іон у кислому середовищі в залежності від умов може
відновлюватися до різноманітних продуктів:
У сильно кислих розчинах йодат окиснює йодиди або йод до I1+
.
Ця реакція потребує присутності таких аніонів як хлориди, броміди,
ціаніди, які стабілізують продукт, що утворюється:
2I- + IO3
- + 3Cl- + 6H+
→ 3ICl + 3H2O
2I2 + IO3
- + 5Cl- + 6H+
→ 5ICl + 3H2O
ICl + Cl- → ICl2
-
Кінцеву точку титрування в залежності від конкретних умов можна
визначати різними способами:
а) надлишкова крапля KIO3 у присутності калію йодиду утворює I2, який з
крохмалем утворює комплекс, забарвлений в інтенсивно-синій колір;
б) в сильно кислому середовищі, необхідному для утворення ICl2
-, крохмаль, як
індикатор, не спрацьовує. В цьому випадку до реакційної суміші додають
невелику кількість органічного розчинника, який не змішується з водою
(CHCl3, C6H6). Після додавання чергової порції калію йодату суміш збовтують.
Титрування ведуть до зникнення червоно-фіолетового забарвлення органічного
шару.
Чутливість такого способу визначення кінцевої точки титрування
співвідносна зі способом, в якому застосовується крохмаль, однак, при
титруванні з крохмалем значно скорочується час титрування.
Реакція окиснення йодидів калію йодатом є зручним джерелом
отримання відомих кількостей йоду (на кожний моль йодату виділяється шість
еквівалентів йоду) і може бути застосована для різноманітних аналітичних
цілей:
Розчини калію йодату хімічно стійкі і можуть зберігатися протягом
тривалого часу.
IO3
-
+ 5I
-
+ 6H+ 3 I2 + 3H2O
IO3
-
+ 6H+ + 5e I
o
+ 3H2O
IO3
-
+6H
+
+ 6e I
-
+ 3H2O
109.
110
Визначення кислоти аскорбінової.Визначення ґрунтується на відновних
властивостях кислоти аскорбінової. Титрантом-окисником є калію йодат в
присутності калію йодиду:
Е=1/2М.м.
Надлишок титрованого розчину калію йодату в кислому середовищі
окиснює калію йодид:
KIO3 + 5KI + 6HCl → 3I2 + 6KCl + 3H2O
Йод, що виділився, забарвлює крохмаль в синій колір.
Методика. Близько 0,5 г субстанції (точна наважка) розчиняють у воді в мірній
колбі ємністю 50 мл, доводять об’єм розчину водою до мітки та перемішують.
До 10 мл приготованого розчину додають 0,5 мл 1% розчину калію йодиду,
2 мл розчину крохмалю та 1 мл 2% розчину хлороводневої кислоти і титрують
0,1 моль/л розчином калію йодату до появи стійкого синього забарвлення.
1 мл 0,1 моль/л розчину калію йодату відповідає 0,008806 г кислоти
аскорбінової, якої в субстанції повинно бути не менш 99,0%.
Розрахунок кількісного вмісту кислоти аскорбінової у відсотках
проводять за формулою 3.7.
3.2.6.5. Нітритометрія
Нітритометрія – титриметричний метод аналізу, який ґрунтується на
окисно-відновних властивостях системи HNO2/NO, Eo
=0,99 B. Редокс-потенціал
системи достатньо великий, тому нітритометрично можна визначати цілий ряд
відновників (As2O3, FeSO4).
Але найчастіше нітритометрію застосовують для кількісного визначення
органічних лікарських речовин, які мають у своєму складі первинну чи
вторинну ароматичні аміногрупи, або нітрогрупу, яку перед визначенням
відновлюють до аміногрупи. Як титрант застосовують натрію нітрит, з якого у
кислому середовищі виділяється нітритна кислота. У загальному вигляді
реакцію діазотування можна представити таким чином:
З наведеного рівняння видно, що в ньому беруть участь 2 молекули
кислоти, з яких одна йде на утворення нітритної кислоти, а інша – солі
діазонію. Однак, для того, щоб реакція проходила стехіометрично, необхідна
+
ArNH2 + 2HCl + NaNO2 [Ar N N] Cl
-
+ NaCl + 2H2O
O
O
OHOH
H
CH
CH2OH
HO
3 + KIO3
3
O
O
H
CH
CH2OH
HO
O O
+ KI + 3H2O
110.
111
присутність надлишку мінеральноїкислоти. Тому на практиці беруть 2,5-3,0
еквівалента кислоти. У присутності надлишку кислоти підвищується також
стійкість діазосполук.
Швидкість реакції утворення діазосполук залежить від природи аміну й
аніону мінеральної кислоти, яка приймає участь у реакції. Аміни, які містять в
ароматичному ядрі елекронноакцепторні замісники (–NO2, –SO3H, –COOH,
–Cl), діазотуються швидше ніж аміни, які містять електроннодонорні замісники
(–OCH3, –CH3 та ін.).
У сірчанокислому середовищі швидкість діазотування менша, ніж у
солянокислому. Підвищується швидкість діазотування у присутності бромід-
іонів, тому до реакційного середовища додають калію бромід, який виконує
роль каталізатору. Більшість діазосполук є нестійкі; їх розкладання
прискорюється при підвищенні температури. А оскільки діазотування – процес
екзотермічний, перед початком реакції розчин, як правило, охолоджують
до 0-10о
С. Однак, деякі діазосполуки досить стійкі й реакцію їх визначення
можна проводити при кімнатній температурі.
Момент еквівалентності визначають за допомогою зовнішніх, внутрішніх
індикаторів або електрометрично (потенціометричне титрування). Як зовнішній
індикатор застосовують йодкрохмальний папір, тобто фільтрувальний папір,
змочений розчином крохмалю та йодиду калію. Як внутрішні індикатори
застосовують тропеолін-00, нейтральний червоний або змішані індикатори,
наприклад, тропеолін-00 у суміші з метиленовим синім. Титрування з
тропеоліном-00 проводять від червоного забарвлення до жовтого, зі змішаним –
від червоно-фіолетового до блакитного.
Титрування з йодкрохмальним папірцем проводять до тих пір, доки
крапля розчину, що титрується, взята через 1 хвилину після додавання розчину
натрію нітриту негайно викликатиме посиніння. При цьому протікає реакція:
2KI + 2NaNO2 + 4HCl → I2+ 2NO↑ + 2KCl + 2NaCl + 2H2O
Аби усунути індикаторну помилку в нітритометрії майже завжди
паралельно проводять контрольний дослід.
Хоча кількість електронів, які віддає первинний ароматичний амін під час
діазотування більша від одного, оскільки реакція протікає стехіометрично і
один моль аміну реагує з одним молем натрію нітриту, прийнято вважати, що
еквівалентна маса дорівнює молекулярній масі.
Метод нітритометричного титрування широко застосовується для аналізу
лікарських речовин, які містять первинну та вторинну аміногрупи (новокаїн,
анестезин, дикаїн, стрептоцид, норсульфазол, та ін.), ацильовану аміногрупу
(фенацетин, парацетамол – після гідролізу), а також нітрогрупу, яку перед
визначенням відновлюють до аміногрупи (левоміцетин).
111.
112
Визначення новокаїну. Воснові визначення лежить реакція:
Е=М.м.
Методика. Близько 0,3 г субстанції (точна наважка) розчиняють в суміші 10 мл
води та 10 мл розведеної хлороводневої кислоти і додають воду до загального
об’єму 80 мл. До одержаного розчину додають 1 г броміду калію і при
постійному перемішуванні титрують 0,1 моль/л розчином нітриту натрію,
додаючи його спочатку зі швидкістю 2 мл за хвилину, а в кінці титрування (за
0,5 мл до еквівалентної кількості) по 0,05 мл кожну хвилину. Точку
еквівалентності визначають за допомогою внутрішнього індикатору
тропеоліну-00 в суміші з метиленовим синім. Титрування проводять при
температурі не вище 18-20о
С. Перехід забарвлення від червоно-фіолетового до
блакитного.
1 мл 0,1 моль/л розчину нітриту натрію відповідає 0,02728 г новокаїну,
якого в субстанції повинно бути не менш 99,5%.
Розрахунок кількісного вмісту новокаїну у відсотках проводять за
формулою 3.10.
Визначення парацетамолу. Для того, щоб нітритометрично визначити
парацетамол спочатку проводять його кислотний гідроліз, а потім п-
амінофенол, що утворився, титрують нітритом натрію:
Е=М.м.
Методика. Близько 0,25 г субстанції (точна наважка) вміщують в конічну
колбу ємністю 100 мл, додають 10 мл розведеної хлороводневої кислоти та
кип’ятять зі зворотним холодильником протягом 1 години. Після цього
Cl
-
+ NaCl + 2H2O
. HCl. HCl
+ NaNO2 + 2HCl
+
NH2
COOCH2CH2N
C2H5
C2H5
N N
COOCH2CH2N
C2H5
C2H5
+ CH3COOH+ H2O
[H+]
OH
NHCOCH3
OH
NH2
Cl
-
+ NaCl + 2H2O
+
+ NaNO2 + 2HCl
NH2
OH
N N
OH
112.
113
холодильник промивають 30мл води, вміст колби кількісно переносять у
склянку для діазотування, промивають колбу 30 мл води і переносять її також в
склянку для діазотування. Потім додають 1 г броміду калію і титрують
0,1 моль/л розчином натрію нітриту, додаючи його спочатку зі швидкістю 2 мл
за 1 хвилину, а в кінці титрування (за 0,5 мл до еквівалентної кількості) по
0,05мл кожну хвилину. Кінець титрування встановлюють за йодкрохмальним
папірцем.
Для цього після додавання чергової порції розчину нітриту натрію й
ретельного перемішування через 3 хвилини беруть скляною паличкою краплю
розчину, що титрується, і наносять на йодкрохмальний папір.
Титрують до тих пір, поки чергова крапля реакційної суміші, нанесеної на
йодкрахмальний папір, не буде негайно викликати появу синього кольору на
папері.
Паралельно проводять контрольний дослід.
1 мл 0,1 моль/л розчину натрію нітриту відповідає 0,01512 г
парацетамолу, якого в субстанції повинно бути не менш 98,5%.
Розрахунок кількісного вмісту парацетамолу у відсотках проводять за
формулою 3.10.
Визначення левоміцетину. Нітритометричне визначення левоміцетину
проводять після відновлення ароматичної нітрогрупи до аміногрупи:
Е=М.м.
Методика. Близько 0,5 г левоміцетину (точна наважка) вміщують в конічну
колбу ємністю 200-250 мл, додають 20 мл концентрованої хлороводневої
кислоти і обережно, невеликими порціями, 5 г цинкового пилу. Потім додають
ще 10 мл концентрованої хлороводневої кислоти, обмиваючи стінки колби, і
після повного розчинення цинкового пилу (допускається легке підігрівання),
розчин кількісно переносять в склянку для діазотування, охолоджують льодом,
додають 3 г калію броміду і повільно титрують 0,1 моль/л натрію нітритом.
Титрування вважають закінченим, коли крапля рідини, що титрується, взята
через 3 хвилини після додавання розчину нітриту натрію буде викликати
негайне посиніння йодкрахмального папірця.
1 мл 0,1 моль/л розчину натрію нітриту відповідає 0,03231 г
левоміцетину, якого в субстанції повинно бути не менш 98,5%.
H
HCl
Zn
H
H
+ NaNO2 + 2HCl
H
+
Cl
-
+
+ 2NaCl + 2H2O
O2N CH
OH
CH CH2OH
N C CHCl2
O
H2N CH
OH
CH CH2OH
N C
O
CH3
H2N CH
OH
CH CH2OH
N C
O
CH3
NN CH
OH
CH CH2OH
N C
O
CH3
113.
114
Розрахунок кількісного вмістулевоміцетину у відсотках проводять за
формулою 3.5.
3.2.6.6. Дихроматометрія
Дихромат калію є більш слабким окисником, ніж KMnO4 або Ce4+
. Однак,
не дивлячись на це, він все ж застосовується в аналізі лікарських речовин
(кількісне визначення гліцерину, етилового спирту в хлороформі). Перевагою
дихромату калію є стійкість його розчинів та можливість отримувати
кристалічний K2Cr2O7 високого ступеня чистоти, що дозволяє готувати
титровані розчини за точною наважкою.
В кислому середовищі дихромат-іон відновлюється до Сr+3
:
Ео
= 1,33 В
Забарвлення розчину K2Cr2O7 недостатньо інтенсивне для визначення за
ним кінцевої точки титрування. Вибір редокс-індикаторів, які можна
використати для дихроматометричного титрування також обмежений
(дифеніламіносульфокислота). Як правило, для повного окиснення речовини
дихроматом потрібен певний час. Враховуючи все це, частіше застосовують
зворотне титрування.
Титровані розчини дихромату калію також використовують для
кількісного визначення лікарських речовин, які утворюють з ним нерозчинні
сполуки (метиленовий синій).
Визначення метиленового синього. В основі визначення лежить реакція
утворення нерозчинного осаду з розчином калію дихромату, надлишок якого
визначають йодометрично:
Е=1/3М.м.
Методика. Близько 0,2 г субстанції (точна наважка) вміщують у склянку
ємністю 100 мл, розчиняють в 40 мл води, нагрітої до 75о
С і доливають 25 мл
0,1 моль/л розчину дихромату калію. Реакційну суміш добре перемішують та
нагрівають протягом 5 хвилин, занурюючи склянку в водяний обігрівач,
Cr2O7
2-
+ 14H+ + 6e 2Cr+3 + 7H2O
2 Cl
-
K2Cr2O7
H2SO4
N
S
+
(H3C)2N N(CH3)2
2
K2Cr2O7 + 6KI + 7H2SO4 Cr2(SO4)3 + 3I2 + 4K2SO4 + 7H2O
I2 + 2Na2S2O3 2NaI + Na2S4O6
Cr2O7
2- + K2SO4 + 2HCl
N
S
+
(H3C)2N N(CH3)2
114.
115
розігрітий до 75о
С.Потім дають охолонути і осад, що випав, відфільтровують
на скляний фільтр № 3. Склянку та фільтр промивають льодяною водою (4 рази
по 2,5 мл), кожного разу повністю відсмоктуючи воду. До фільтрату (з
промивними водами) додають 150 мл води, 30 мл розведеної сульфатної
кислоти та 2 г йодиду калію, добре перемішують, залишають на 5 хвилин. Йод,
що виділився, титрують 0,1 моль/л розчином натрію тіосульфату, додаючи в
кінці титрування 2 мл розчину крохмалю.
Паралельно проводять контрольний дослід.
1 мл 0,1 моль/л розчину калію дихромату відповідає 0,01066 г
метиленового синього, якого в перерахунку на суху речовину повинно бути не
менш 97,0%.
Розрахунок кількісного вмісту метиленового синього у відсотках
проводять за формулою:
,
)100(
100100)(
%
Bm
TKVV
X к
−⋅
⋅⋅⋅⋅−
= (3.15)
де V – об’єм 0,1 моль/л розчину натрію тіосульфату, витраченого на
титрування в прямому досліді, мл;
Vк – об’єм 0,1 моль/л розчину натрію тіосульфату, витраченого на
титрування у контрольному досліді, мл;
К – коефіцієнт поправки до молярності 0,1 моль/л розчину натрію
тіосульфату;
Т – титр 0,1 моль/л розчину натрію тіосульфату за метиленовим синім,
г/мл;
m – маса наважки метиленового синього, г;
В – процент вологості метиленового синього.
Визначення етилового спирту в хлороформі. В основі визначення лежить
реакція окиснення етилового спирту калію дихроматом, надлишок якого
визначають йодометрично:
3C2H5OH + 2K2Cr2O7 + 16HNO3 → 3CH3COOH + 4Cr(NO3)3 + 4KNO3 + 11H2O
K2Cr2O7 + 6KI + 14HNO3 → 2Cr(NO3)3 + 3I2 + 8KNO3 + 7H2O
I2 + 2Na2S2O3 → 2NaI + Na2S4O6
Методика. В склянку з притертою пробкою ємністю 300-500 мл вносять 25 мл
0,1 моль/л калію дихромату, 25 мл концентрованої нітратної кислоти і
охолоджують у льодяній воді. До охолодженої суміші додають 1 мл субстанції і
залишають на 5 хвилин, періодично перемішуючи. Потім додають 100 мл води,
5 мл розчину калію йодиду, залишають на 5 хв у темному місці і йод, що
виділився, титрують 0,1 моль/л розчином натрію тіосульфату (індикатор –
крохмаль).
Паралельно проводять контрольний дослід.
Е=1/4М.м.; 1 мл 0,1 моль/л розчину калію дихромату відповідає 0,00115 г
етилового спирту, якого повинно бути не менш 0,6-1%.
115.
116
Розрахунок кількісного вмістуетилового спирту у відсотках проводять за
формулою:
,
100)(
%
.. анд
к
V
TKVV
X
⋅⋅⋅−
= (3.16)
де Vк – об’єм 0,1 моль/л розчину натрію тіосульфату, витраченого на
титрування у контрольному досліді, мл;
V – об’єм 0,1 моль/л розчину натрію тіосульфату, витраченого на
титрування в прямому досліді, мл;
Vд. ан. – об’єм хлороформу, взятий для аналізу, мл;
К – коефіцієнт поправки до молярності 0,1 моль/л розчину натрію
тіосульфату;
Т – титр 0,1 моль/л розчину натрію тіосульфату за етанолом, г/мл;
3.2.6.7. Цериметрія
Метод ґрунтується на окисненні лікарських речовин-відновників
церієм (IV):
Як титрант застосовують розчин сульфату церію (IV) в присутності
сульфатної кислоти, який за окисною здатністю наближається до розчину
перманганату калію (електродний потенціал Ce4+
в 1 моль/л сульфатній кислоті
дорівнює + 1,44). Сірчанокислі розчини церію (IV) дуже стійкі, реакція завжди
приводить до утворення Ce3+
.
Для фіксування точки еквівалентності можна застосовувати ряд окисно-
відновних індикаторів (дифеніламін, фероїн) або визначати її фізико-хімічними
методами. Особливо зручні у використанні різноманітні 1,10-фенантрометани,
потенціал переходу забарвлення яких відповідає потенціалу в точці
еквівалентності основної реакції.
Цериметрію можна застосовувати для визначення широкого кола
лікарських речовин (броміди, розчин перекису водню, кислота аскорбінова,
токоферол, вікасол, аміназин).
Визначення вікасолу. Спочатку взаємодією з натрію гідроксидом осаджують
2-метил-1,4-нафтохінон, який потім відновлюють цинком в присутності
хлороводневої кислоти і титрують 2-метил-1,4-нафтогідрохінон, що утворився,
0,1 моль/л розчином сульфату церію:
+1e
Ce4+ Ce3+
O
O
CH3
O
O
CH3
SO3Na
NaOH + Na2SO3 + H2O
116.
117
Е=1/2М.м.
Індикатор – фероїн,який одержують розчиненням о-фенантроліну в
1,48% розчині феруму(II) сульфату. В момент еквівалентності Ce4+
окиснює
Fe2+
до Fe3+
, який з о-фенантроліном дає комплекс блакитного кольору.
Титрують до появи зеленого забарвлення (блакитний колір комплексу з жовтим
розчином титранту дає зелене забарвлення):
Методика. Близько 0,3 г субстанції (точна наважка) розчиняють у 20 мл води,
переносять в ділильну лійку, додають 5 мл 1 моль/л розчину натрію гідроксиду
та екстрагують хлороформом (3 рази по 20 мл). Об’єднаний хлороформний
екстракт промивають 10 мл води, фільтрують через паперовий фільтр,
змочений хлороформом і промивають фільтр 5 мл хлороформу. Хлороформ
відганяють насухо у вакуумі при кімнатній температурі. Залишок розчиняють у
15 мл льодяної оцтової кислоти, додають 15 мл розведеної хлороводневої
кислоти, 3 г цинкового пилу і залишають на 30 хвилин в темному місці, зрідка
перемішуючи. Потім вміст колби швидко фільтрують через вату в іншу колбу.
Осад в колбі та фільтр негайно промивають водою (3 рази по 10 мл). До
одержаного фільтрату додають 2-3 краплі розчину о-фенантроліну і титрують
0,1 моль/л розчином сульфату церію до появи зеленого забарвлення.
Паралельно проводять контрольний дослід.
1 мл 0,1 моль/л розчину сульфату церію відповідає 0,01652 г вікасолу,
якого в субстанції повинно бути не менш 95,0%.
Розрахунок кількісного вмісту вікасолу у відсотках проводять за
формулою 3.10.
3+
3
Fe + Ce3++ Ce4+
2+
Fe
3
N
N
N
N
O
CH3
OOH
OH
CH3
OH
OH
CH3
O
O
CH3
+ Ce2(SO4)3 + H2SO4+ 2Ce(SO4)2
HCl
Zn




![6
1. Визначення тотожності лікарських речовин
1.1. Ідентифікація неорганічних іонів
Визначення тотожності неорганічних лікарських речовин у більшості
випадків зводиться до ідентифікації окремих іонів – аніонів та катіонів, що
входять до їх складу.
В цьому розділі наведено реакції ідентифікації катіонів та аніонів, а
також деяких органічних аніонів, які входять до складу лікарських речовин.
Методики викладено відповідно до вимог діючої науково-технічної
документації. Наведено також нефармакопейні реакції, які часто
використовуються в фармацевтичному аналізі.
1.1.1. Катіони
1.1.1.1. Амоній NH4
+
1. 1 мл розчину солі амонію (0,002-0,006 г іона амонію) нагрівають з 0,5 мл
розчину натрію гідроксиду; виділяється амоніак, який виявляють за запахом
та посинінням вологого червоного лакмусового папірця:
2. 0,01-0,02 г солі амонію розчиняють в 1 краплі води, додають 1 краплю
реактиву Несслера; утворюється осад червоно-бурого кольору:
1.1.1.2. Бісмут Bi3+
1. Лікарські засоби бісмуту (близько 0,05 г іона бісмуту) збовтують з 3 мл
розведеної хлороводневої кислоти і фільтрують. До фільтрату додають 1 мл
розчину натрію сульфіду або сірководню; утворюється коричнево-чорний
осад, який розчиняється при додаванні рівного об’єму концентрованої
нітратної кислоти.
2Bi3+
+ 3S2-
→ Bi2S3↓
Bi2S3 + 8HNO3 → 2Bi(NO3)3 + 2NO↑ + 3S↓ + 4H2O
2. Лікарські засоби бісмуту (близько 0,05 г іона бісмуту) збовтують з 5 мл
розведеної сульфатної кислоти і фільтрують. До фільтрату додають 2 краплі
розчину калію йодиду; утворюється чорний осад, що розчиняється в
надлишку реактиву; з’являється жовтувато-оранжеве забарвлення:
Bi3+
+ 3I- → BiI3↓
↓ BiI3 + I- → [BiI4]-
NH4
+
+ OH
-
NH4OH NH3 + H2O
NH4
+
+ 2HgI4
2-
+ 2OH
-
NH2
Hg
Hg
I
I
+
I
-
+ 5I
-
+ 2H2O](https://image.slidesharecdn.com/1-170625000218/85/1-5-320.jpg)
![7
1.1.1.3. Ферум (II) Fe2+
1. До 2 мл розчину солі феруму (II) (близько 0,02 г іона феруму) додають
0,5 мл розведеної хлороводневої кислоти та 1 мл розчину калію фериціаніду;
утворюється синій осад, який не розчиняється у мінеральних кислотах, але
руйнується лугами з утворенням гідроксиду феруму (II):
3Fe2+
+ 2[Fe(CN)6]3-
→ Fe3[Fe(CN)6]2↓
Fe3[Fe(CN)6]2 + 6OH- → 3Fe(OH)2↓ + 2[Fe(CN)6]3-
Можливо також утворення сполуки KFe[Fe(CN)6].
2. До розчину солі феруму (II) (близько 0,02 г іона феруму) додають розчин
амонію сульфіду; утворюється чорний осад, розчинний у розведених
мінеральних кислотах:
Fe2+
+ S2-
→ FeS↓
1.1.1.4. Ферум (III) Fe3+
1. До 2 мл розчину солі феруму (III) (близько 0,02 г іона феруму) додають
0,5 мл розведеної хлороводневої кислоти та 1-2 краплі розчину калію
фероціаніду; утворюється синій осад:
4Fe3+
+ 3[Fe(CN)6]4-
→ Fe4[Fe(CN)6]3↓
Можливо також утворення осаду KFe[Fe(CN)6]. Осад розкладається лугами:
↓Fe4[Fe(CN)6]3 + 12OH- → 4Fe(OH)3↓ + 3[Fe(CN)6]4-
2. До 2 мл розчину солі феруму (III) (близько 0,001 г іона феруму) додають
0,5 мл розведеної хлороводневої кислоти та 1-2 краплі розчину амонію
роданіду; з’являється червоне забарвлення:
Fe3+
+ 3SCN- → Fe(SCN)3
3. До розчину солі феруму (III) (близько 0,001 г іона феруму) додають розчин
амонію сульфіду; утворюється чорний осад, розчинний у розведених
мінеральних кислотах:
2Fe3+
+ 3S2-
→ Fe2S3↓
1.1.1.5. Калій K+
1. До 2 мл розчину солі калію (0,01 – 0,02 г іона калію) додають 1 мл розчину
винної кислоти, 1 мл розчину натрію ацетату, 0,5 мл 95% спирту, збовтують;
поступово утворюється білий кристалічний осад, розчинний у розведених
мінеральних кислотах і розчинах лугів:](https://image.slidesharecdn.com/1-170625000218/85/1-6-320.jpg)
![8
Утворенню осаду сприяє охолодження реакційної суміші та потирання
скляною паличкою стінок пробірки.
2. До 2 мл розчину солі калію (0,005-0,01 г іона калію), попередньо прожареної
для видалення солей амонію, додають 0,5 мл розведеної оцтової кислоти та
0,5 мл розчину натрію кобальтинітриту; утворюється жовтий кристалічний
осад:
2K+
+ Na+
+ [Co(NO2)6]3-
→ K2Na[Co(NO2)6]↓
Реакцію не можна проводити в лужному або сильнокислому середовищі
внаслідок руйнування комплексного йону:
[Co(NO2)6]3-
+ 3OH- → Co(OH)3↓ + 6NO2
-
[Co(NO2)6]3-
+ 6H+
→ Co3+
+ 6HNO2
3. Сіль калію, внесена в безбарвне полум’я, забарвлює його в фіолетовий колір
або при розгляданні крізь синє скло – в пурпурово-червоний.
1.1.1.6. Кальцій Ca2+
1. До 1 мл розчину солі кальцію (0,002-0,02 г іона кальцію) додають 1 мл
розчину амонію оксалату; утворюється білий осад, нерозчинний у
розведеній оцтовій кислоті та розчині амоніаку, розчинний у розведених
мінеральних кислотах:
Ca2+
+ C2O4
2-
→ CaC2O4↓
2. Сіль кальцію, змочена хлороводневою кислотою і внесена у безбарвне
полум’я забарвлює його в цегляно-червоний колір.
COOH
CH
CH
COOH
OH
OH
CH
CH
COOK
COOH
OH
OH
+ K
+ + H
+
H
+
+ CH3COONa CH3COOH + Na
+
CH
CH
COOH
OH
OH
COOH
CH
CH
COOH
OH
OH
COOK
+ H
+
+ K
+
CH
CH
COOH
OH
OH
COOK
CH
CH
OH
OH
COOK
COO
-
+ H2O+ OH
-](https://image.slidesharecdn.com/1-170625000218/85/1-7-320.jpg)
![9
1.1.1.7. Магній Mg2+
1. До 1 мл розчину солі магнію (0,002-0,005 г іона магнію) додають 1 мл
розчину амонію хлориду, 1 мл розчину амоніаку та 0,5 мл розчину натрію
фосфату; утворюється білий кристалічний осад, розчинний у розведених
мінеральних кислотах та оцтовій кислоті:
Mg2+
+ NH4
+
+ PO4
3-
→ MgNH4PO4↓
↓MgNH4PO4 + H+
→ Mg2+
+ NH4
+
+ HPO4
2-
Амонію хлорид додається для запобігання утворення в реакційній суміші
осаду гідроксиду магнію. Надлишку амонію хлориду слід уникати, оскільки
можуть утворюватися розчинні комплекси [MgCl3]-, [MgCl4]2-.
1.1.1.8. Арсен As3+
1. До 0,3 мл розчину солі тривалентного арсену (близько 0,03 г іона арсеніту)
додають 0,5 мл розведеної хлороводневої кислоти та 2 краплі розчину
натрію сульфіду або сірководню; утворюється жовтий осад, нерозчинний в
концентрованій хлороводневій кислоті, розчинний у розчині амоніаку:
2As3+
+ 3S2-
→ As2S3↓
↓As2S3 + OH- → AsS3
3-
+ AsO3
3-
+ 3H2O
2. До 0,3 мл розчину солі тривалентного арсену (близько 0,003 г іона арсеніту)
додають 1 – 2 краплі розчину аргентуму нітрату; утворюється жовтий осад,
розчинний у розведеній нітратній кислоті та розчині амоніаку:
AsO3
3-
+ 3Ag+
→ Ag3AsO3↓
↓Ag3AsO3 + 6NH3 → 3[Ag(NH3)2]+
+ AsO3
3-
1.1.1.9. Арсен As5+
1. До 0,3 мл розчину солі п’ятивалентного арсену (близько 0,03 г іона
арсенату) додають 0,5 мл розведеної хлороводневої кислоти, 2 краплі
розчину натрію сульфіду або сірководню і нагрівають; утворюється жовтий
осад сульфіду арсену, нерозчинний у концентрованій хлороводневій кислоті,
розчинний у розчині амоніаку:
AsO4
3-
+ 2H+
+ S2-
→ AsO3
3-
+ S↓ +H2O
2AsO3
3-
+ 12H+
+ 3S2-
→ As2S3↓ + 6H2O
2. До 0,3 мл розчину солі п’ятивалентного арсену (близько 0,001 г іона
арсенату) додають 1–2 краплі розчину аргентуму нітрату; утворюється
коричневий осад, розчинний у розведеній нітратній кислоті та розчині
амоніаку:
AsO4
3-
+ 3Ag+
→ Ag3AsO4↓
↓Ag3AsO4 + 6NH3 → 3[Ag(NH3)2]+
+ AsO4
3-
3. До 0,3 мл розчину солі п’ятивалентного арсену (близько 0,001 г іона
арсенату) додають по 1 мл розчинів амонію хлориду, амоніаку та магнію](https://image.slidesharecdn.com/1-170625000218/85/1-8-320.jpg)
![10
сульфату; утворюється білий кристалічний осад розчинний у розведеній
хлороводневій кислоті (відмінність від арсенітів):
AsO4
3-
+ Mg2+
+ NH4
+
→ MgNH4AsO4↓
↓MgNH4AsO4 + H+
→ Mg2+
+ NH4
+
+ HAsO4
2-
1.1.1.10. Натрій Na+
1. 1 мл розчину солі натрію (0,01-0,03 г іона натрію), підкислюють розведеною
оцтовою кислотою, якщо необхідно, фільтрують, потім додають 0,5 мл
розчину цинк-уранілацетату; утворюється жовтий кристалічний осад:
Na+
+ Zn2+
+[(UO2)3(CH3COO)8]2-
+ CH3COO-
+ 9H2O →
→ NaZn[(UO2)3(CH3COO)9] . 9H2O↓
2. Сіль натрію, змочена хлороводневою кислотою, при внесенні в безбарвне
полум’я забарвлює його в жовтий колір.
1.1.1.11. Меркурій (II) Hg2+
1. До 2 мл розчину солі меркурію (II) (близько 0,05 г іона меркурію) додають
0,5 мл розчину натрію гідроксиду; утворюється жовтий осад:
Hg2+
+ 2OH- → HgO↓ + H2O
2. До 1 мл розчину солі меркурію (II) (0,01-0,03 г іона меркурію) обережно по
краплі додають розчин калію йодиду; утворюється червоний осад, який
розчиняється у надлишку реактиву:
Hg2+
+2I- → HgI2↓
↓HgI2 + 2I- → [HgI4]2-
1.1.1.12. Меркурій (I) Hg2
2+
1. Солі меркурію (I) утворюють з розчинами лугів чорний осад оксиду
меркурію (I):
Hg2
2+
+ 2OH- → Hg2O↓ + H2O
2. Солі меркурію (I) утворюють з розчинами йодидів жовто-зелений осад
меркурію (I) йодиду:
Hg2
2+
+ 2І- → Hg2І2↓
Осад розчиняється в надлишку реактиву з виділенням металічної ртуті:
Hg2І2↓ + 2І- → [HgI4]2-
+ Hg↓
3. З розчинами хлоридів солі меркурію (I) утворюють білий осад меркурію (І)
хлориду:
Hg2
2+
+ 2Cl- → Hg2Cl2↓](https://image.slidesharecdn.com/1-170625000218/85/1-9-320.jpg)
![11
4. При взаємодії солей меркурію(І) з розчином амоніаку утворюється темний
осад вільної ртуті:
Hg2Cl2 + 2NH3 →HgNH2Cl + Hg↓ + NH4Cl
1.1.1.13. Цинк Zn2+
1. До 2 мл нейтрального розчину солі цинку (0,005-0,02 г іона цинку) додають
0,5 мл розчину натрію сульфіду або сірководню; утворюється білий осад,
нерозчинний у розведеній оцтовій кислоті та легко розчинний у розведеній
хлороводневій кислоті:
Zn2+
+ S2-
→ ZnS↓
↓ZnS + 2H+
→ Zn2+
+ H2S↑
2. До 2 мл розчину солі цинку (0,005 – 0,02 г іона цинку) додають 0,5 мл
розчину калію фероціаніду; утворюється білий осад, нерозчинний у
розведеній хлороводневій кислоті:
3Zn2+
+ 2K+
+2[Fe(CN)6]4-
→ K2Zn3[Fe(CN)6]2↓
3. При взаємодії розчину солі цинку з розчином натрію гідроксиду
утворюється білий драглеподібний осад, розчинний у надлишку реактиву з
утворенням цинкатів:
Zn2+
+ 2OH- → Zn(OH)2↓
↓Zn(OH)2 + 2OH- → [Zn(OH)4]2-
1.1.1.14. Аргентум Ag+
1. До 1 мл розчину солі аргентуму (близько 0,005 г іона аргентуму) додають
розчин амоніаку до розчинення осаду, що утворюється спочатку, потім 2-3
краплі розчину формальдегіду і нагрівають; на стінках пробірки
утворюється блискучий наліт або випадає чорний осад металічного срібла:
2Ag+
+ 2OH- → Ag2O↓ + H2O
Ag2O + 4NH3 + H2O → 2[Ag(NH3)2]OH
2[Ag(NH3)2]OH + HCOH → 2Ag↓ + HCOO- + NH4
+
+ 3NH3↑ + H2O
2. 0,001 – 0,002 г іона аргентуму розчиняють в 1-2 краплях води, додають
1 краплю розведеної хлорводневої кислоти або розчину хлориду натрію;
утворюється білий сирнистий осад:
Ag+
+ Cl- → AgCl↓
Осад нерозчинний в кислотах, але легко розчиняється у розчинах амоніаку і
тіосульфату натрію:
↓AgCl + 2NH3 → [Ag(NH3)2]+
+ Сl-
↓AgCl + 2S2O3
2-
→ [Ag(S2O3)2]3-
+ Cl-](https://image.slidesharecdn.com/1-170625000218/85/1-10-320.jpg)
![12
1.1.1.15. Купрум Cu2+
1. 0,002 – 0,005 г солі купруму розчиняють в 1 – 2 краплях води, додають 1
краплю розчину амоніаку; утворюється блакитний осад, який розчиняється у
надлишку розчину амоніаку; з’являється темно-синє забарвлення:
2Cu2+
+ 2OH- + SO4
2-
→ Cu2(OH)2SO4↓
↓Cu2(OH)2SO4 + 4NH4
+
+ 2OH- → 2[Cu(NH3)4]2+
+ SO4
2-
+ 4H2O
1.1.2. Аніони
1.1.2.1. Ацетати CH3COO-
1. 2 мл розчину ацетату (0,02 – 0,06 г іона ацетату) нагрівають з рівною
кількістю концентрованої сульфатної кислоти та 0,5 мл 95% спирту;
відчувається запах етилацетату:
2. До 2 мл нейтрального розчину ацетату (0,02 - 0,06 г іона ацетату) додають
0,2 мл розчину феруму (III) хлориду; з’являється червоно-буре забарвлення,
яке зникає від додавання розведених мінеральних кислот:
6CH3COO- + 3Fe3+
+ 2H2O → [Fe3(CH3COO)6(OH)2]+
+ 2H+
1.1.2.2. Бензоати C6H5COO-
1. До 2 мл нейтрального розчину бензоату (0,01 – 0,02 г іона бензоату) додають
0,2 мл розчину феруму (III) хлориду; утворюється рожево-жовтий осад,
розчинний в ефірі:
Реакцію проводять в нейтральному середовищі, бо в лужному феруму (III)
хлорид утворює бурий осад гідроксиду феруму (III), а в кислому комплексна
сіль розчиняється.
2. З розчином купруму сульфату нейтральні розчини бензоатів утворюють осад
бірюзового кольору:
CH3COOH + C2H5OH CH3C
O
OC2H5 + H2O
H2SO4
6 + 2Fe3+ + 10H2O
3
-
Fe3+ . Fe(OH)3
. 7H2O +
+ 3
COO
-
COO
COOH
2 + Cu2+
2
-
Cu2+
COO
-
COO](https://image.slidesharecdn.com/1-170625000218/85/1-11-320.jpg)
![13
1.1.2.3. Броміди Br-
1. До 1 мл розчину броміду (0,002 – 0,03 г іона броміду) додають 2 мл
розведеної хлороводневої кислоти, 0,5 мл розчину хлораміну, 1 мл
хлороформу та збовтують, хлороформний шар забарвлюється в жовто-бурий
колір:
2. До 2 мл розчину броміду (0,002 – 0,01 г іона броміду) додають 0,5 мл
розведеної нітратної кислоти та 0,5 мл розчину аргентуму нітрату;
утворюється жовтуватий сирнистий осад, нерозчинний у розведеній
нітратній кислоті та важко розчинний у розчині амоніаку:
Br- + Ag+
→ AgBr↓
↓AgBr + 2NH3 → [Ag(NH3)2]+
+ Br-
3. До 0,002 – 0,005 г броміду додають 1 краплю розчину купруму сульфату,
5 крапель концентрованої сульфатної кислоти; з’являється чорний осад; від
додавання декількох крапель води відбувається знебарвлення:
Cu2+
+ 2Br- → CuBr2↓
1.1.2.4. Йодиди I-
1. До 2 мл розчину йодиду (0,002-0,01 г іона йодиду) додають 0,5 мл
розведеної нітратної кислоти та 0,5 мл розчину аргентуму нітрату;
утворюється жовтий сирнистий осад, нерозчинний у розведеній нітратній
кислоті та розчині амоніаку:
I- + Ag+
→ AgI↓
2. До 2 мл розчину йодиду (0,003–0,02 г іона йодиду) додають 0,2 мл
розведеної сульфатної кислоти, 0,2 мл розчину натрію нітриту або розчину
феруму (III) хлориду та 2 мл хлороформу; при збовтуванні хлороформний
шар забарвлюється в фіолетовий колір:
2I- + 2NO2
- + 4H+
→ I2 + 2NO↑ + 2H2O
2I- + 2Fe3+
→ I2 +2Fe2+
3. При нагріванні 0,1 г йодиду з 1 мл концентрованої сульфатної кислоти
виділяються фіолетові пари йоду.
+ Cl
-
+ Na++ 2Br
-
+ 2H+ Br2 +
SO2N
Na
Cl
SO2NH2](https://image.slidesharecdn.com/1-170625000218/85/1-12-320.jpg)

![15
приливають 5 – 6 крапель концентрованої сульфатної кислоти, так аби
рідини не змішалися; на межі двох рідин з’являється буре кільце:
6Fe2+
+ 2NO3
- + 8H+
→ 6Fe3+
+ 2NO + 4H2O
FeSO4 + NO → [FeNO]SO4
1.1.2.7. Нітрити NO2
-
1. До лікарського засобу (близько 0,001 г іона нітриту) додають 2 краплі
розчину дифеніламіну; з’являється синє забарвлення (див. нітрати, 1.1.2.6.1).
2. До лікарського засобу (близько 0,03 г іона нітриту) додають 1 мл розведеної
сульфатної кислоти; віділяються жовто-бурі пари (відмінність від нітратів).
2NO2
- + 2H+
→ HNO2 → NO↑ + NO2↑ + H2O
2NO + O2 → 2NO2↑
3. Декілька кристалів антипірину розчиняють у фарфоровій чашці в 2 краплях
розведеної хлороводневої кислоти, додають 2 краплі розчину нітриту
(близько 0,001 г іона нітриту); з’являється зелене забарвлення (відмінність
від нітратів):
4. До 4-5 крапель розчину нітриту додають таку саму кількість розведеної
сульфатної кислоти і по краплі приливають розчин калію перманганату;
відбувається знебарвлення розчину калію перманганату:
5NO2
- + 2MnO4
- + 6H+
→ 5NO3
- + 2Mn2+
+ 3H2O
5. Так само як і нітрати, нітрити дають реакцію з феруму (II) сульфатом (див.
нітрати, 1.1.2.6.4). На відміну від нітратів реакція може протікати в
слабокислому середовищі.
1.1.2.8. Саліцилати C6H4(OH)COO-
1. До 2 мл нейтрального розчину саліцилату (0,002-0,01 г іона саліцилату)
додають 2 краплі розчину феруму (III) хлориду; з’являється синьо-фіолетове
або червоно-фіолетове забарвлення, яке зберігається при додаванні
невеликої кількості розведеної оцтової кислоти, але зникає при додаванні
розведеної хлороводневої кислоти. При цьому утворюється білий
кристалічний осад саліцилової кислоти:
+ H2O+ NO2
-
+ H+
N
N
CH3
CH3
O
C6H5
ON
N
N
CH3
CH3
H
O
C6H5
C
ONa
O
OH FeO
O
O
C
+ NaCl + HClCl
-
+
FeCl3+](https://image.slidesharecdn.com/1-170625000218/85/1-14-320.jpg)

![17
1.1.2.11. Тартрати (HOCHCOO-)2
1. До 1 мл розчину тартрату (близько 0,02 г іона тартрату) додають кристалик
калію хлориду, 0,5 мл 95% спирту; утворюється білий кристалічний осад,
розчинний у розведених мінеральних кислотах та розчинах лугів.Реакцію
проводять у присутності натрію ацетату при охолодженні та потиранні
скляною паличкою стінок пробірки (див. калій, 1.1.1.5.1).
2. 0,25 мл розчину тартрату (близько 0,005 г іона тартрату) нагрівають з 1 мл
концентрованої сульфатної кислоти та декількома кристалами резорцину;
через 15-30 с з’являється вишнево-червоне забарвлення.
1.1.2.12. Фосфати PO4
3-
1. До 1 мл розчину фосфату (0,01-0,03 г іона фосфату), нейтралізованого до pH
близько 7,0, додають декілька крапель розчину аргентуму нітрату;
утворюється жовтий осад, розчинний у розведеній нітратній кислоті та
розчині амоніаку:
PO4
3-
+ 3Ag+
→ Ag3PO4↓
↓Ag3PO4 + 3H+
→ 3Ag+
+ H3PO4
↓Ag3PO4 + 6NH3 → 3[Ag(NH3)2]+
+ PO4
3-
2. До 1 мл розчину фосфату (0,01-0,03 г іона фосфату) додають 1 мл розчину
амонію хлориду, 1 мл розчину амоніаку та 0,5 мл розчину магнію сульфату;
утворюється білий кристалічний осад, розчинний у розведених мінеральних
кислотах (див. магній, 1.1.1.7.1).
3. До 1 мл розчину фосфату (0,01-0,03 г іона фосфату) в розведеній нітратній
кислоті додають 2 мл розчину амонію молібдату і нагрівають; утворюється
жовтий кристалічний осад, розчинний у розчині амоніаку:
PO4
3-
+ 3NH4
+
+ 12MoO4
2-
+ 24H+
→ (NH4)3PO4
. 12MoO3↓ + 12H2O
1.1.2.13. Хлориди Cl-
1. До 2 мл розчину хлориду (0,002-0,01 г іону хлориду) додають 0,5 мл
розведеної нітратної кислоти і 0,5 мл розчину аргентуму нітрату;
утворюється білий сирнистий осад нерозчинний у розведеній нітратній
кислоті і розчинний у розчині амоніаку:
Для солей органічних основ дослідження розчинності осаду аргентуму
хлориду проводять після відфільтровування і промивання осаду водою
(якщо цього не зробити може утворитися осад органічної основи).
[Ag(NH3)2]Cl + H2O+ 2NH4OHAgCl
Cl
-
+ Ag+ AgCl](https://image.slidesharecdn.com/1-170625000218/85/1-16-320.jpg)
![18
1.1.2.14. Цитрати
1. До 1 мл нейтрального розчину цитрату (0,002-0,01 г іона цитрату) додають
1 мл розчину кальцію хлориду; розчин залишається прозорим; при
кип’ятінні утворюється білий осад, розчинний у розведеній хлороводневій
кислоті:
2. До лікарського засобу (0,001-0,002) г іона цитрату додають 0,5 мл оцтового
ангідриду і нагрівають; через 20-40 с з’являється червоне забарвлення.
3. До 3-5 крапель розчину цитрату (0,0001 г іона цитрату) додають 3-5 крапель
0,01 моль/л розчину калію перманганату, 3-5 крапель насиченої бромної
води і злегка підігрівають. Після охолодження надлишок брому і оксид
марганцю (IV), що іноді утворюється, руйнують додаванням твердої
сульфосаліцилової кислоти. Випадає білий кристалічний осад. Таку саму
реакцію дають тартрати, а її проведенню заважають феноли і ароматичні
аміни, які утворюють осади з бромною водою:
1.2. Ідентифікація органічних лікарських речовин
Аналіз органічних лікарських речовин відрізняється від аналізу
неорганічних сполук. Молекула органічної речовини складається з основи
(скелету) (вуглеводневий (аліфатичний) ланцюг, ароматична або
гетероциклічна структура) і певного набору функціональних груп, наявність,
розташування та взаємний вплив яких і визначають хімічні й фармакологічні
C
CH2
CH2
COO
-
COO
-
COO
-
HO
2
Ca3
2+
3-
C
CH2
CH2
HO
COO
COO
COO
tº
+ 3Ca2+C
CH2
CH2
COO
-
COO
-
COO
-
2HO
+ 5Br2 2 C
CHBr2
CHBr3
OCO2 + 5HBr +C
CH2
CH2
COOH
COOH
O
+ CO2 + H2OC
CH2
CH2
COOH
COOH
O+ [O]C
CH2
CH2
HO
COOH
COOH
COOH](https://image.slidesharecdn.com/1-170625000218/85/1-17-320.jpg)
![19
властивості сполуки. Тому аналіз лікарських речовин органічної природи
зводиться до реакцій, направлених на визначення функціональних груп.
1.2.1. Ненасичені вуглеводні
Характерними реакціями ненасичених вуглеводнів є, головним чином,
реакції електрофільного приєднання:
1. При взаємодії лікарських речовин, які містять ненасичений зв’язок, з
бромною водою спостерігається знебарвлення останньої (наприклад, при
проведенні реакції талеохінної спроби на хінін).
Використання в такій реакції як середовища безводного розчинника,
наприклад, тетрахлорметану, дає змогу відрізнити алкени чи алкіни від
ароматичних сполук, які реагують з бромом з виділенням бромоводню (він не
розчиняється в тетрахлорметані й виділяється у вигляді бульбашок).
Методика. Розчин 1 г (0,3 мл) брому в 100 мл чотирихлористого вуглецю
додають краплями до розчину 50 мг речовини в 2 мл чотирихлористого
вуглецю. Розчин брому одразу знебарвлюється.
2. Калію перманганат в нейтральному або слаболужному середовищі
окиснює ненасичені сполуки, при цьому його розчини знебарвлюються:
В кислому середовищі, особливо при нагріванні, реакція не зупиняється
на стадії окиснення подвійного зв’язку і проходить далі з розривом вуглець-
вуглецевого зв’язку з утворенням відповідних альдегідів або, навіть, кислот:
Реакція не є специфічною, тому в фармацевтичному аналізі широкого
застосування не знаходить. Використовується вона, наприклад, для визначення
домішок цінаммоїлкокаїну й інших відновлюючих речовин у кокаїну
гідрохлориді.
В літературі описано використання цієї реакції для ідентифікації цинку
ундеценоату (а) та кислоти коричної (б).
Методика. а) 0,1 г цинку ундеценоату розчиняють у суміші 2 мл 1 моль/л
сульфатної кислоти і 5 мл льодяної оцтової кислоти (речовина не розчиняється
у воді) і додають по краплі 0,25 мл розчину калію перманганату –
спостерігається знебарвлення розчину переманганату калію;
б) 0,1 г коричної кислоти нагрівають з 0,1 г калію перманганату і 5 мл
1 моль/л сульфатної кислоти – утворюється бензальдегід, який визначають за
запахом.
3. π – Зв’язки ненасичених вуглеводнів можуть брати участь в утворенні
комплексів з солями важких металів, наприклад, стибію (III) хлоридом.
C C + [O] + H2O C C
OHOH
H
C C
OHO
+ [O] H2O + 2 C O](https://image.slidesharecdn.com/1-170625000218/85/1-18-320.jpg)

![21
3. Наявність атомів йоду визначають за появою фіолетових парів йоду під
час піролізу речовини (йодоформ) або окиснення її при нагріванні з розведеною
нітратною (хініофон) чи концентрованою сульфатною кислотою (білігност,
йодогност, сергозин).
4. Фторорганічні сполуки ідентифікують після мінералізації з сумішшю
для спікання. До розчину, що містить фторид-іони додають розчин кальцію
хлориду – з’являється біла каламуть (фторафур):
2F- + CaCl2 → CaF2↓ + 2Cl-
Більш сучасним є метод спалення у колбі з киснем. Продукти реакції
розчиняють у воді і додають до розчину феруму роданіду; спостерігається
значне послаблення забарвлення (фторурацил):
Fe(SCN)3 + 6F- → [FeF6]3- + 3SCN-
1.2.3. Спиртовий гідроксил
1. Найбільш загальною реакцією на спирти є реакція етерифікації.
Взаємодія спиртів з ангідридами, хлорангідридами або карбоновими кислотами
в присутності концентрованої сульфатної кислоти приводить до утворення
складних ефірів:
Продукти реакції ідентифікують за характерним запахом або
температурою плавлення.
Методика. а) 2 мл етанолу змішують з 0,5 мл льодяної оцтової кислоти, 1 мл
концентрованої сульфатної кислоти і нагрівають до кипіння – з’являється
характерний запах етилацетату;
б) до 0,1 г метиландростендіолу додають 0,6 мл оцтового ангідриду,
4,5 мл безводного піридину і нагрівають на водяному обігрівачі при 50-60°С
протягом трьох годин у колбі зі зворотним холодильником; до охолодженої
рідини додають 30 мл крижаної води. Через 30 хв осад відфільтровують,
переосаджують водою з ацетону, висушують і визначають температуру
плавлення отриманого моноацетату;
в) розчиняють 0,2 г ментолу в 0,5 мл безводного піридину, додають 3 мл
15% розчину хлорангідриду 3,5 динітробензойної кислоти в безводному
піридині і нагрівають на водяному обігрівачі протягом 10 хв; до отриманого
ROH +
RO H +
RO H +
O
C
C
R'
R'
O
O
C
O
R'RO + R'COOH
C R'
O
Cl C
O
R'RO + HCl
HO
O
R'C
H2SO4
C
O
R'RO + H2O](https://image.slidesharecdn.com/1-170625000218/85/1-20-320.jpg)
![22
розчину додають 7 мл води і витримують у кризі протягом 30 хв. Осад
відфільтровують, промивають крижаною водою, перекристалізовують з
ацетону і визначають температуру плавлення.
2. Для ідентифікації спиртів широко застосовують реакції окиснення.
Утворені при цьому альдегіди або кетони встановлюють за характерним
запахом або відповідними якісними реакціями:
Ця реакція застосовується для підтвердження тотожності ефедрину
гідрохлориду, етанолу, бензилового спирту, а також для визначення домішки
метилового спирту в етиловому.
Методика. а) 0,05 г ефедрину гідрохлориду розчиняють в 1 мл води, додають
кристал калію фериціаніду і нагрівають до кипіння – з’являється запах
бензальдегіду:
б) до 0,1 мл бензилового спирту додають 5 мл 3% розчину калію
перманганату і 1 мл 1 моль/л сульфатної кислоти; утворюється бензальдегід,
який визначають за характерним запахом;
в) до 0,5 мл етанолу додають 4,5 мл води і 2 мл розчину калію
перманганату в фосфатній кислоті. Через 10 хв по краплі додають насичений
розчин натрію гідросульфіту до знебарвлення розчину, 1 мл 2% розчину
динатрієвої солі хромотропової кислоти, поступово, 10 мл концентрованої
сульфатної кислоти (порціями по 1 мл через 1 хв) і перемішують. Поява
фіолетового забарвлення свідчить про наявність домішки метилового спирту:
R CH2 OH
[O]
CR
H
O
CH
OH
R R'
[O]
CR R'
O
K3[Fe(CN)6]
C
H
O
. HCl
CH3NHOH
CH3CHCH
H2SO4 (конц.)
5CH3OH + 2KMnO4 + 3H3PO4 5H C
O
H
+ 2MnHPO4 + K2HPO4 + 8H2O
HO
+ H C
O
H
+
H2O
HO
SO3Na
SO3Na
NaO3S
OH
OH
NaO3S](https://image.slidesharecdn.com/1-170625000218/85/1-21-320.jpg)
![23
Однією з відомих реакцій на спирти, зокрема етанол, є реакція утворення
йодоформу:
Методика. До 0,5 мл етанолу додають 5 мл розчину натрію гідроксиду і 2 мл
0,1 моль/л розчину йоду – з’являється специфічний запах і поступово
утворюється жовтий осад йодоформу. Осад може здаватися аморфним, однак
під мікроскопом можна розпізнати гексагональні і зірчасті кристали.
Йодоформна реакція не є специфічною на спирти, оскільки сполуки, що
містять етоксильну (-OC2H5), ацетильну (-CO-CH3), моно і дийодацетильні
групи також дають позитивну спробу. В реакцію утворення йодоформу
вступають етанол, хлоралгідрат, оцтовий альдегід, метилкетони (наприклад,
ацетон) і вторинні спирти, які при окисненні утворюють метилкетони.
1.2.4. Багатоатомні спирти
1. Характерною властивістю багатоатомних спиртів є їх здатність
утворювати комплексні сполуки. Наприклад, при взаємодії з борною кислотою
утворюються комплексні кислоти і нейтральний або слаболужний, спочатку,
розчин стає кислим, що визначають за допомогою кислотно-основних
індикаторів:
Методика. 0,05 г речовини, що аналізується, розчиняють в 2 мл води, додають
краплю 0,02 моль/л розчину натрію гідроксиду і краплю розчину
фенолфталеїну. В іншій пробірці в 2 мл води розчиняють 0,5 г натрію
C
C
OH
OH
H
H
R
R'
+ H3BO32
C
C
H
H
R
R'
O
O
B
O
O C
C
R
R'
H
H
H
+
HO
[O]
HO
CHHO
SO3H
CH2
SO3H
HO3S
OH
OH
HO3S
SO3H
HO
SO3H
OH
HO3S
O
HO3S
R CH
OH
CH3 + NaOI
R C
O
CH3
+ NaI + H2OR C
O
CH3
+ 3NaOI R C
O
CI3 + 3NaOH
R C
O
CI3 + NaOH R C
O
ONa + CHI3](https://image.slidesharecdn.com/1-170625000218/85/1-22-320.jpg)


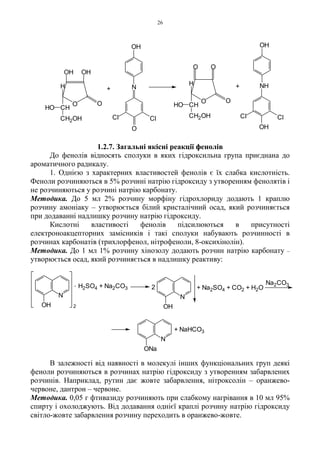

![28
Методика. До 0,25 г синестролу додають 1 мл оцтового ангідриду і 2мл
безводного піридину. Кип’ятять зі зворотним холодильником протягом 15 хв,
охолоджують, додають 50 мл води і ретельно струшують. Осад
відфільтровують, промивають водою, сушать при 100-105оС. Температура
плавлення виділеного діацетату синестролу складає 137-139оС.
5. Особливістю фенолів у порівнянні зі спиртами є здатність досить легко
окиснюватися під дією різноманітних окисників. В результаті утворюються
продукти, які мають характерне забарвлення.
При змочуванні декількох крупинок апоморфіну гідрохлориду 1 краплею
нітратної кислоти з’являється криваво-червоне забарвлення.
6. Хінони, які утворюються при окисленні фенолів конденсуються з
амінами з утворенням хінонімінів і продуктів їх конденсації. Прикладом такої
реакції є реакція індофенолової спроби.
Методика. 0,05 г речовини, що досліджується, розчиняють в 0,5 мл розчину
амоніаку і додають 3-4 краплі розчину хлораміну (хлорного вапна, натрію
гіпохлориту, бромної води) суміш нагрівають на водяному обігрівачі – через
декілька хвилин з’являється забарвлення, яке змінюється від додавання
кислоти:
Реакцію окиснення в присутності амоніаку дають не лише феноли але й
деякі їх прості ефіри. Прикладом може слугувати реакція талеохінної спроби.
Методика. До 5 мл 0,1% розчину хініну гідрохлориду додають 2-3 краплі
бромної води і 1 мл розчину амоніаку – з’являється зелене забарвлення:
індофенол (синє забарвлення)
H C6H4OH
H2O
NH3[O]
O
O
O
NH
O
N OHOH
N
CH CH2
Br2
N
CH CH2
OH OH
N
CH CH2
Br Br
4NH4OH
N
H3CO
CH
OH
N
N
O
O CH
OH
CH
OHNH
NH](https://image.slidesharecdn.com/1-170625000218/85/1-27-320.jpg)


![31
а) Реакція “срібного дзеркала”. Реактивом є амоніачний розчин оксиду
аргентуму (реактив Тойленса). В результаті реакції альдегід окислюється до
кислоти, а окис аргентуму відновлюється до металічного срібла:
Методика. В добре вимиту пробірку наливають розчин аргентуму нітрату і
краплями додають розчин амоніаку до розчинення осаду, що утворився
спочатку. Приливають 2-3 краплі розчину альдегіду і обережно нагрівають на
водяному обігрівачі – виділяється металічне срібло у вигляді дзеркала чи сірого
осаду.
б) Реакція з реактивом Фелінга.
Методика. До розчину 0,2 г глюкози в 5 мл води додають 10 мл реактиву
Фелінга і нагрівають до кипіння – випадає цеглинно-червоний осад:
в) Реакція з реактивом Несслера. Альдегіди відновлюють реактив
Несслера з утворенням осаду металічної ртуті:
Реакція з реактивом Несслера є більш чутливою, ніж реакції з реактивами
Тойленса і Фелінга, і тому її застосовують для відкриття домішок альдегідів в
інших речовинах, наприклад, у діетиловому ефірі.
Разом з тим треба пам’ятати, що ці реакції не є специфічними, їх дають
будь які відновники – ендіоли, о-, п-дифеноли, похідні гідразину і т. ін.
Альдегіди і кетони
1. Реакції приєднання. Серед реакцій приєднання слід зупинитися на
реакціях з бісульфітом натрію. Альдегіди і кетони реагують з ним і дають
бісульфітні похідні, які добре кристалізуються:
C
O
H
R + 2[Ag(NH3)2]OH C
O
R
ONH4
+ 2Ag + 3NH3 + H2O
C
O
H
R + K2HgI4 + 3KOH R COOK + 4KI + Hg + 2H2O
C
O
H
R + NaHSO3 C
H
R
OH
SO3Na
HOHC
HOHC
NaOOC
OOC
CHOH
CHOH
COOK
COO
Cu
C
C
C
C
C
CH2OH
OH
OH
OH
HO
H
H
H
H
H
O
2+ + 4
2CuOH
t Cu2O + H2O
3
COONa
C
C
COOK
H
H
OH
OH
NaOH
C
C
C
C
CH2OH
OH
OH
OH
H
H
H
HO
H
COONa
2KOH
+2CuOH +2H2O](https://image.slidesharecdn.com/1-170625000218/85/1-30-320.jpg)

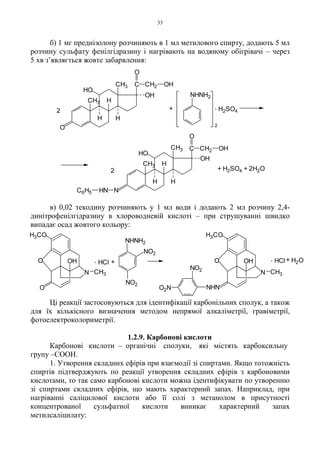





![39
3. Первинні аліфатичні й ароматичні аміни, а також вторинні аміни
можна розрізнити за допомогою реакції з нітритною кислотою. Первинні аміни
в реакції з нітритною кислотою утворюють діазосполуки. Але, якщо ароматичні
діазосполуки стійкі, то аліфатичні – нестабільні і одразу після утворення, навіть
на холоду, швидко розкладаються з виділенням азоту:
R–NH2 + ONOH → R–OH + H2O + N2↑
Загальна методика виглядає так: в мікропробірці змішують декілька
міліграмів натрію нітриту з хлороводневокислим розчином аміну. Інтенсивне
виділення безбарвного газу – азоту свідчить про наявність у сполуки первинної
аліфатичної аміногрупи.
В кислому середовищі сам нітрит натрію розкладається з утворенням
окислів азоту. При уважному розгляданні видно, що над шаром рідини
збирається бурий газ:
NaNO2 + HCl → NaCl + HNO2
2HNO2 → H2O + NO↑ + NO2↑
Методика. Розчиняють 0,1 г мексилену гідрохлориду [(RS)-1-метил-2-(2,6-
ксилілокси)-етиламіну гідрохлориду] в 3 мл 0,02 моль/л хлороводневої кислоти
і додають декілька кристалів натрію нітриту – спостерігається бурхливе
виділення газу.
Вторинні аміни при взаємодії з нітритною кислотою утворюють
нітрозаміни:
Продуктом реакції найчастіше є жовта важка рідина або кристалічний
осад, розчинні в діетиловому ефірі. Ефірний шар відділяють і випаровують до
сухого залишка. Отриманий нітрозамін при нагріванні з концентрованою
сульфатною кислотою гідролізується з утворенням нітритної кислоти, яка в
присутності фенолу дає реакцію Лібермана (див. феноли, 1.2.7.6).
Первинні ароматичні аміни при взаємодії в кислому середовищі з
азотистою кислотою утворюють безбарвні або блідо-жовті солі діазонію, які з
лужними розчинами фенолів дають реакцію азосполучення з утворенням
азобарвників.
Методика. 0,05 г анестезину розчиняють в 2 мл води, підкисленої 3 краплями
розведеної хлороводневої кислоти, додають 3 краплі 0,1 моль/л розчину натрію
нітриту і збовтують; отриманий розчин додають до 3 мл лужного розчину β-
нафтолу – з’являється вишнево-червоне забарвлення або утворюється
оранжево-червоний осад:
NH + HONO
R
R1
N NO
R
R1
+ H2O](https://image.slidesharecdn.com/1-170625000218/85/1-38-320.jpg)


![42
Реакція не є специфічною для вторинних амінів, в літературі описано її
використання для підтвердження тотожності прокаїну гідрохлориду
(новокаїну).
8. Для виявлення третинних амінів рекомендується реакція з цитриновою
кислотою. Розчини цитринової, аконітової, малонової кислот в оцтовому
ангідриді при нагріванні з третинними амінами набувають червоного або
фіолетового забарвлення.
Методика. До 0,5 мл 2% розчину лимонної кислоти в оцтовому ангідриді
додають слідову кількість або краплю спиртового розчину аміну і нагрівають
на водяному обігрівачі – з’являється червоне або фіолетове забарвлення.
Позитивну реакцію дають аліфатичні, аліциклічні, мішані, ароматичні і
аліциклічні, а також третинні ароматичні аміни. Солі органічних і неорганічних
кислот з лужними і лужно-земельними металами заважають реакції на відміну
від інших солей. Оскільки реакція дуже чутлива рекомендується паралельно
проводити контрольний дослід. Очевидно через неспецифічність широкого
застосування в фармацевтичному аналізі вона не знайшла, хоча й
рекомендується для підтвердження тотожності ціметідину (2-ціано-1-метил-3-
[2-(5-метилімідазол-4-іл-метилтіо)-етил]-гуанідину).
9. Наявність неподіленої пари електронів надає більшості азотовмісних
органічних сполук не тільки основних властивостей, але й здатність
утворювати комплекси і тому для ідентифікації різноманітних аліфатичних,
аліциклічних, гетероциклічних і мішаних первинних, вторинних, третинних
амінів у фармацевтичному аналізі широко застосовують реакції зі специфічною
групою комплексоутворювачів – загальноалкалоїдними осадовими реактивами.
Загальноалкалоїдні осадові реактиви
1. Реактив Люголя, Вагнера, Бушарда (розчин йоду в калію йодиді).
2. Реактив Драгендофа (розчин бісмуту йодиду в калію йодиді).
3. Реактив Майера (розчин меркурію йодиду в калію йодиді).
4. Реактив Марме (розчин кадмію йодиду в калію йодиді).
5. Реактив Зонненштейна – фосфорномолібденова кислота
(H3PO4
.12MoO3
.2H2O).
6. Реактив Шейблера – фосфорновольфрамова кислота
(H3PO4
.12WoO3
.2H2O).
7. Реактив Бертрана – кремнійвольфрамова кислота (SiO2
.12WoO3
.4H2O).
C2H5OH
KOH
+ +
. HNO3
++
N
O
O
H
HNO3
. HCl
H
CO(CH2)2O N
CH3
CH3
N C4H9
CO(CH2)2O N
CH3
CH3
N C4H9
N
O
O
CH3
CH3
NO CO(CH2)2
N
O
O
C4H9N
O
N
OK](https://image.slidesharecdn.com/1-170625000218/85/1-41-320.jpg)
![43
8. 5% Розчин таніну (використовують свіжий розчин).
9. Насичений розчин пікринової кислоти.
1.2.14. Піридиновий цикл
Серед азотовмісних гетероциклічних сполук значну групу складають
похідні піридину і тому піридиновий цикл слід відзначити як специфічну
функціональну групу.
1. Найпростішою реакцією на піридиновий цикл є реакція піролізу. При
нагріванні нікотинової кислоти, її солей, нікотинаміду з безводним карбонатом
натрію відчувається запах піридину (див. карбонові кислоти, 1.2.9.3).
2. Для ідентифікації похідних піридину, у яких вільне α, α'-положення,
проводять реакцію утворення поліметинової основи з 2,4-динітрохлорбензолом.
Методика. До 0,01-0,02 г нікотинової кислоти додають 0,05 г 2,4-
динітрохлорбензолу, 3 мл 95% етилового спирту і кип’ятять протягом 1
хвилини – утворюється сіль піридинію. При додаванні 2 крапель 10% розчину
натрію гідроксиду відбувається розмикання піридинового циклу і утворюється
похідне глутаконового альдегіду - поліметинова основа, що забарвлює розчин в
буро-червоний колір. Далі в результаті гідролізу червоне забарвлення розчину
поступово зникає:
Поліметинові основи утворюються і при використанні таких реагентів як
тіоціанат брому (роданбромідний реактив), тіоціанат хлору, ціанід брому,
хлороформ, хлоралгідрат. В присутності зазначених реагентів в лужному
середовищі відбувається розмикання піридинового циклу і утворюється
глутаконовий альдегід. При наступному додаванні первинних ароматичних
амінів (анілін, новокаїн, сульфацил-натрій) відбувається їх конденсація з
глутаконовим альдегідом і утворюються основи Шиффа забарвлені в жовтий,
оранжевий або червоний колір.
Методика. До 2 мл бромної води додають 2-3 краплі розчину амонію роданіду
(до знебарвлення). В отриманому розчині розчиняють 0,02 г речовини, що
H2O
[OH]
+
CH
HO
C O
H
C O
H
Cl
-
+
+
N
COOH NO2
Cl
NO2
COOH
N
NO2
NO2
NH
COOH
COOH
NO2
NO2
NO2
NO2
NH2](https://image.slidesharecdn.com/1-170625000218/85/1-42-320.jpg)








![53
4. Для ідентифікації гідразонів застосовують реакцію гідролізу, під час
якого утворюється відповідний альдегід, котрий можна ідентифікувати за
характерним запахом.
Методика. 0,05 г фтивазиду нагрівають з 10 мл розведеної хлороводневої
кислоти – з’являється сильний запах ваніліну:
1.2.19. Тіоли, тіони, тіоефіри, тіоаміди
До цієї групи відносять сполуки, які містять функціональні групи:
1. Найбільш загальною реакцією, яка використовується для ідентифікації
сірковмісних сполук є реакція окисної мінералізації з наступною
ідентифікацією утвореного сульфат-іона взаємодією з барію хлоридом.
Методика. а) До 0,2 г етоксиду (N,N`-[ди-(п-етоксифеніл)]тіосечовини)
додають 5 мл розведеної нітратної кислоти і доводять до кипіння, потім
охолоджують, фільтрують через беззольний фільтр. Фільтрат дає характерну
реакцію на сульфати;
б) до 20 мг пропілтіоурацилу додають 8 мл бромної води і перемішують
протягом 5 хв. Розчин кип’ятять до знебарвлення, дають охолонути і
фільтрують. Додають 2 мл розчину барію хлориду – виникає білий осад.
2.Для ідентифікації сірковмісних сполук зі ступенем окиснення сірки (-2),
застосовують реакції кислотного або лужного гідролізу. При цьому
утворюється сірководень або сульфіди, що їх підкисленням переводять у
сірководень, який ідентифікують за запахом або реакцією з плюмбуму
ацетатом.
Методика. а) 0,1 г тіофосфаміду вміщують у пробірку, додають 3 мл
розведеної хлороводневої кислоти і кип’ятять у полум’ї пальника –
відчувається запах сірководню;
;;; R C NHR'
S
R S R'R C R'
S
R SH
+ NH2NH2
. 2HCl+
N
COOH
H2O
N
CONHN CH
OCH3
OH
OH
OCH3
C
H
O
HCl, tº
N(CH3)2
N(CH3)2
N(CH3)2
N(CH3)2CHN
N CH
Cl
-
+
HCl
CHN
NH CH](https://image.slidesharecdn.com/1-170625000218/85/1-52-320.jpg)
![54
б) кип’ятять 0,1 г моносульфіраму (тетраетилтіоурацилу моносульфід) з 2
моль/л розчином хлороводневої кислоти – пари, що виділяються, забарвлюють
фільтрувальний папір, змочений розчином плюмбуму ацетату, в чорний колір;
в) 0,05 г метіоніну нагрівають у пробірці з 5-6 краплями 30% розчину
натрію гідроксиду до отримання сплаву. До охолодженого сплаву додають 5 мл
води і підкислюють розведеною сульфатною кислотою – з’являється запах
сірководню і меркаптану:
г) 0,2 г тіопентал-натрію розчиняють в 5 мл розчину натрію гідроксиду,
додають 2 мл розчину ацетату плюмбуму і кип’ятять – випадає темний осад.
Після охолодження і підкислення концентрованою хлороводневою кислотою
виділяється сірководень, який ідентифікують за запахом і потемнінням
фільтрувального паперу, змоченого розчином ацетату плюмбуму:
3. Одним з реактивів, що широко застосовується для ідентифікації тіолів
є нітропрусид натрію [Na2Fe(NO)(CN)5].
Методика. а) 0,05 г меркаптопурину вміщують у пробірку, розчиняють у
0,5 мл розчину натрію гідроксиду, розбавляють водою до 2 мл і додають 0,5 мл
свіжоприготованого розчину натрію нітропрусиду; пробірку струшують –
з’являється жовто-зелене забарвлення, що переходить при підкисленні
розведеною хлороводневою кислотою в темно-зелене;
б) 0,02 г мерказолілу розчиняють у 1 мл води, додають 1 мл розчину
натрію гідроксиду і збовтують. До отриманого розчину додають 3 краплі
розчину натрію нітропрусиду – через декілька хвилин з’являється жовте
+ 2NH3 + 2Na2CO3 + 2CH3COONa
HC
CH3
CH2
CH
CH2 CH3
COONa
H5C2
+PbS
N
N
NaS
H O
O
C2H5
CH
CH3
CH2 CH2 CH3 + 6NaOH + (CH3COO)2Pb
чорний
+ CH3SNa + Na2S + CH3OH + 2NH3
+CH2 CH2 CH C
ONH2
2HO
ONa
Na2S + CH3SNa + 2H2SO4 H2S + CH3SH + Na2SO4 + NaHSO4
+ 5NaOH2H3C S CH2 CH2 CH C
O
OH
NH2
H2S + PbCl2PbS + 2HCl](https://image.slidesharecdn.com/1-170625000218/85/1-53-320.jpg)


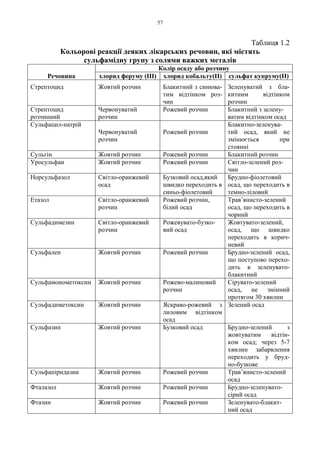





![63
2.3.2. Дослідження на сульфати
Розчини сульфатів, у залежності від їх концентрації, утворюють з
розчинами солей барію (BaCl2, Ba(NO3)2) білий осад або муть, які не зникають
від додавання розведеної хлороводневої кислоти:
Реакцію проводять у середовищі розведеної хлороводневої кислоти, щоб
запобігти утворенню осадів BaCO3 або BaHPO4.
Гранична чутливість реакції – 0,003 мг сульфат-іона в 1 мл розчину.
Методика. До 10 мл розчину лікарського засобу, приготовленого за вимогами
монографії, додають 0,5 мл розведеної хлороводневої кислоти, 1 мл розчину
барію хлориду, перемішують і через 10 хвилин порівнюють з еталоном (10 мл),
до якого додано таку ж кількість реактивів, як і в розчин, що досліджується.
2.3.3. Дослідження на солі амонію
У залежності від властивостей лікарської речовини та допустимої
концентрації домішки солей амонію її визначення проводиться одним з двох
методів.
Метод I ґрунтується на здатності солей амонію утворювати з реактивом
Несслера (лужний розчин калію тетрайодмеркурату (II)) жовто-бурий осад або
жовте забарвлення у залежності від концентрації:
Гранична чутливість реакції – 0,0003 мг іона амонію в 1 мг розчину.
Методика. До 10 мл розчину лікарського засобу, приготовленого за вимогами
монографії, додають 0,15 мл реактиву Несслера, перемішують і через 5 хвилин
порівнюють з еталонним розчином (10 мл), що містить таку ж кількість
реактивів, як і розчин, що досліджується.
У лікарських речовинах, які містять лужноземельні та важкі метали,
визначення проводять після розчинення в мінімальній кількості води та
додавання 2 мл розчину натрію гідроксиду та 2 мл розчину натрію карбонату.
Розчин розбавляють водою до необхідної концентрації, збовтують і
фільтрують. Визначення домішки проводять в 10 мл фільтрату за основною
методикою.
В лікарських речовинах, які містять більше 0,03% домішки феруму,
визначення проводять після його зв’язування у безбарвний комплекс з натрію-
калію тартратом:
У цьому випадку визначення проводять після додавання до 10 мл розчину
лікарського засобу 2 крапель розчину натрію гідроксиду та 3 мл 20% розчину
SO4
2- + Ba2+ BaSO4
[FeC4H2O6]+ + 2H+Fe3+ + C4H4O6
2-
+
NH2
Hg
Hg
I
I
NH4
+
+ 2[HgI4]2- + 2OH
- I
-
+ 5I
-
+ 2H2O](https://image.slidesharecdn.com/1-170625000218/85/1-62-320.jpg)

![65
Методика. До 10 мл розчину лікарського засобу, приготовленого за вимогами
монографії, додають 2 мл розчину сульфосаліцилової кислоти та 1 мл розчину
амоніаку і через 5 хвилин порівнюють з еталонним розчином Б іонів феруму
(10 мл), до якого додано таку ж кількість реактивів, як і до розчину, що
досліджується.
У сполуках магнію визначення домішки іонів феруму проводять у
середовищі амоніачного буферу, щоб запобігти утворенню осаду гідроксиду
магнію.
При визначенні домішки феруму в сполуках магнію поступають так само,
як зазначено вище, але перед додаванням розчину амоніаку до розчину
лікарського засобу додають 0,5 мл розчину амонію хлориду.
При визначенні домішок феруму у солях алюмінію застосовують розчин
натрію гідроксиду, в якому гідроксид алюмінію розчиняється.
Визначення солей феруму в сполуках алюмінію проводять таким чином:
до розчину лікарського засобу, приготовленого як зазначено у відповідній
монографії, додають 5 мл розчину сульфосаліцилової кислоти і 2 мл розчину
натрію гідроксиду і порівнюють з еталонним розчином Б іона феруму (10 мл),
до якого додають таку ж кількість реактивів, як і до розчину, що досліджується.
Визначення солей феруму в органічних сполуках проводять після
спалювання наважки лікарського засобу в концентрованій сульфатній кислоті.
Залишок після спалювання обробляють 2 мл концентрованої хлороводневої
кислоти при нагріванні на водяному обігрівачі і додають 2 мл води. Якщо
потрібно, одержаний розчин фільтрують, фільтр промивають 3 мл води. Розчин
разом з промивними водами нейтралізують концентрованим розчином
амоніаку, доводять об’єм розчину водою до 10 мл. Далі чинять, як зазначено
вище.
2.3.6. Дослідження на солі цинку
Домішку іонів цинку визначають по утворенню білого осаду або муті при
взаємодії з розчином калію гексаціаноферату (II):
Гранична чутливість реакції – 0,001 мг цинк-іона в 1 мл розчину.
Методика. До 10 мл розчину лікарського засобу, приготовленого за вимогами
монографії, додають 2 мл хлороводневої кислоти, 5 крапель розчину калію
фероціаніду і через 10 хвилин порівнюють з еталонним розчином Б іона цинку
(10 мл), до якого додають таку ж кількість реактивів, як і до розчину, що
досліджується.
Проведенню досліджень заважає наявність солей феруму (III). Його
вплив усувається додаванням гідроксиду амонію, котрий утворює з солями
феруму осад гідроксиду Fe(OH)3, який відфільтровують. Солі цинку
залишаються у фільтраті у вигляді розчинних цинкатів [Zn(OН)4]2-
.
У разі появи в розчині, що досліджується, синього забарвлення,
необхідно спочатку відділити ферум. Для цього до такого розчину, нагрітого до
3Zn2+ + 2K+ + 2[Fe(CN)6]4- K2Zn3[Fe(CN)6]2](https://image.slidesharecdn.com/1-170625000218/85/1-64-320.jpg)

![67
Мінімальна кількість арсену, яка може бути відкрита цим методом
становить 0,0005 мг.
Методика. У колбу, в якій знаходиться попередньо підготовлена лікарська
речовина, додають від 10 до 12 крапель розчину дихлориду олова, 2 г
гранульованого цинку (який не містить арсену) і відразу закривають колбу
пробкою зі вставленою в неї верхньою частиною приладу. Вміст колби
обережно збовтують і залишають на 1 годину. При цьому температура
реакційної суміші не повинна перевищувати 40оС.
Паралельно в другому такому ж приладі проводять контрольний дослід з
усіма реактивами та додаванням 0,5 мл еталонного розчину арсену. Через
1 годину смужку паперу, оброблену розчином меркурію дихлориду, вміщують
у розчин калію йодиду для більш чіткого проявлення забарвлення та видалення
надлишку меркурію дихлориду:
Через 10 хвилин розчин калію йодиду зливають, смужку паперу ретельно
промивають декілька разів водою методом декантації в тій же склянці та
висушують між листками фільтрувального паперу.
Метод II. Ґрунтується на відновленні сполук арсену до металічного
арсену. Як відновник застосовують розчини натрію гіпофосфіту та
хлороводневої кислоти:
При нагріванні металічний арсен, який утворюється, спостерігають у
вигляді бурого осаду або забарвлення:
Якщо до охолодженої реакційної суміші додати ефір та перемішати,
арсен збирається на межі двох рідин у вигляді бурої плівки.
Метод також застосовують для визначення селену (червоне забарвлення)
та телуру (коричневе забарвлення) або у випадках визначення домішки арсену в
лікарських речовинах, які мають у своєму складі бісмут, меркурій, аргентум,
сульфіди та сульфіти.
Гранична чутливість реакції – 0,01 мг арсену в 10 мл реакційної суміші.
Методика. Наважку лікарської речовини (підготовлену згідно з вимогами
монографії) вносять у пробірку, додають 5 мл розчину натрію гіпофосфіту,
вміщують у киплячий водяний обігрівач і нагрівають протягом 15 хвилин.
В розчині, що досліджується, не повинно спостерігатися ні побуріння, ні
утворення бурого осаду.
У разі побуріння або утворення бурого осаду в пробірку після
охолодження додають 3 мл води, 5 мл ефіру та ретельно збовтують. При
наявності арсену на межі рідин утворюється бура плівка.
HgCl2 + 2KI HgI2 + 2KCl
HgI2 + 2KI K2[HgI4]
NaH2PO2 + HCl H3PO2 + NaCl
As2O3 + 3H3PO2 2As + 3H3PO3
2AsCl3 + 3H3PO2 + 3H2O 2As + 3H3PO3 + 6HCl](https://image.slidesharecdn.com/1-170625000218/85/1-66-320.jpg)








![76
3.2.1.2. Метод Фольгарда базується на осадженні хлоридів, бромідів, йодидів
надлишком стандартного розчину аргентуму нітрату з наступним його
відтитровуванням стандартним розчином роданіду (тіоціанату) амонію.
Як індикатор в методі Фольгарда використовують іон феруму(III), який
вводиться у розчин у вигляді залізо-амонієвого галуну.
Застосування феруму(III) як індикатора базується на його здатності
утворювати з роданід-іонами у водних розчинах комплексну сполуку криваво-
червоного кольору:
Іони аргентуму утворюють з роданід-іонами важкорозчинну сполуку:
Розрахунки показують, що утворення комплексу феруму з роданід-іонами
почнеться тільки після повного осадження катіонів аргентуму у вигляді
аргентуму роданіду, а потім зайва крапля розчину амонію роданіду буде
реагувати з іоном феруму(III), забарвлюючи розчин у червоний колір.
Умови титрування:
1. Середовище повинно бути кислим. Це необхідно для пригнічення гідролізу
іону феруму(III), оскільки індикатор – це сіль, утворена слабкою основою і
сильною кислотою:
Продукт гідролізу – гідроксид феруму(III) червоно-бурого кольору –
заважає точному встановленню точки еквівалентності. Для пригнічення
гідролізу розчини підкислюють нітратною кислотою.
2. При титруванні іонів аргентуму роданідом амонію перша зміна забарвлення
розчину відбувається приблизно за 1% до моменту еквівалентності, що
пов’язано з адсорбцією осадом аргентуму роданіду іонів аргентуму. Для
прискорення їх десорбції з поверхні осаду необхідно енергійне перемішування
розчину у кінці титрування.
З метою економії розчину аргентуму нітрату застосовують непрямий
метод Фольгарда. Точну наважку солі галогеніда розчиняють у воді,
підкислюють нітратною кислотою, додають 1мл розчину залізо-амонієвого
галуну і 0,1 мл 0,1 моль/л розчину амонію роданіду. Виникає криваво-червоне
забарвлення:
FeNH4(SO4)2 + 3NH4SCN → [Fe(SCN)3] + 2(NH4)2SO4
Розчин титрують 0,1 моль/л розчином аргентуму нітрату. Спочатку
реагує галогенід, а після досягнення моменту еквівалентності надлишкова
крапля титранту реагує з феруму (ІІІ) роданідом внаслідок чого розчин
знебарвлюється:
NaBr +AgNO3 → AgBr↓ + NaNO3
3AgNO3 + [Fe(SCN)3] → 3AgSCN↓ + Fe(NO3)3
NH4
+
+ Fe
3+
+ 2SO4
2-
Fe3+
+3H2O Fe(OH)3 + 3H+
FeNH4(SO4)2
Fe3+ + 3SCN
-
[Fe(SCN)3]
Ag+ + SCN
- AgSCN](https://image.slidesharecdn.com/1-170625000218/85/1-75-320.jpg)

![78
3.2.1.3. Метод Фаянса передбачає використання для аргентометричного
титрування адсорбційних індикаторів при визначенні хлорид-, бромід- та
йодид-іонів.
Адсорбційні індикатори – це слабкі органічні кислоти, які дисоціюють за
схемою:
Найчастіше як адсорбційні індикатори використовують флуоресцеїн, еозин,
еозинат натрію, бромфеноловий синій та ін.
У процесі титрування галогенід-іонів іонами аргентуму формуються
осади галогенідів аргентуму, які схильні до утворення колоїдів. Поки не
досягнуто моменту еквівалентності, колоїдні частинки галогенідів аргентуму
адсорбують галогенід-іони, які є у надлишку, і набувають негативного заряду.
Негативно заряджені частинки осаду притягують до себе іони протилежного
заряду, наприклад:
В момент еквівалентності, який співпадає з ізоелектричною точкою, осад
не буде мати заряду.
Перша ж надлишкова краплина розчину аргентуму нітрату створює у
розчині надлишок іонів аргентуму, які починають адсорбуватися на осаді
йодиду аргентуму, надаючи осаду позитивного заряду: ↓[(AgI)Ag+
]NO3
-. Як
протийони поряд з нітрат-іонами будуть також адсорбуватися забарвлені аніони
індикатору: ↓[(AgI)Ag+
]Ind-. Це призведе до зміни забарвлення поверхні осаду.
Зміна забарвлення поверхні осаду свідчить про необхідність припинити
титрування.
Умови титрування:
1. Титрування ведуть при значенні pH, яке визначено для кожного
адсорбційного індикатора, наприклад: з флуоресцеїном в нейтральному або
слабокислому середовищі, з еозином – в кислому, при pH 2.
2. Оскільки адсорбція іонів індикатору відбувається на поверхні осаду, то
вигідно, щоб вона була якомога більшою. Особливо розгалужену поверхню
мають колоїдні розчини. Тому титрування з адсорбційними індикаторами
краще вести, якщо осад хоча б частково знаходиться у вигляді колоїдних
часток.
Щоб запобігти коагуляції колоїдного розчину, до розчину додають
захисні колоїди (антикоагулянти): декстрин або крохмаль.
Методом Фаянса з бромфеноловим синім визначають кількісний вміст
хлоридів, бромідів і йодидів. При спільній присутності титрується вся сума
галогенідів.
Методом Фаянса з еозинатом натрію визначають йодиди. Визначенню не
заважають хлориди, заважають броміди.
H
+
+ Ind
-
HInd
AgI + KNO3AgNO3 + KI [(AgI)I ]K+](https://image.slidesharecdn.com/1-170625000218/85/1-77-320.jpg)



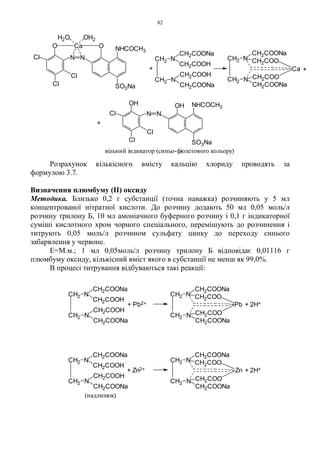
![83
Розрахунок кількісного вмісту плюмбуму (ІІ) оксиду у відсотках
проводять за формулою 3.6.
3.2.3. Меркуріметрія
Меркуріметрія належить до методів комплексонометричного титрування
з використанням неорганічних лігандів. Метод ґрунтується на утворенні
малодисоційованих сполук з катіоном меркурію (II) (Hg2+
).
Меркуріметрія дозволяє визначати лікарські речовини, які містять
галогенід-іони.
Як стандартні розчини, застосовують розчини меркурію (II) нітрату –
Hg(NO3)2 або меркурію (II) перхлорату – Hg(ClO4)2. При цьому відбуваються
такі реакції:
При кількісному визначенні хлоридів та бромідів титруванням розчинами
солей меркурію (II) як індикатори використовують натрію нітропрусид або
дифенілкарбазон.
Натрію нітропрусид [натрій пентаціанід нітрозил феррат (III)] з катіонами
меркурію (II) утворює білий осад:
При використанні дифенілкарбазону забарвлення розчину змінюється від
жовто-червоного до рожево-фіолетового:
Під час визначення йодидів у процесі титрування утворюється
безбарвний комплекс калію тетрайодмеркурату K2HgI4:
N N
C
NHN
C6H5
C6H5C6H5
C6H5
C
NH N
Hg
NN
OO2O C
NH NH
N N
C6H5
C6H5
+ Hg2+
2H+
Сумарно: 4KI + Hg(NO3)2 K2[HgI4] + 2KNO3
2KI + Hg(NO3)2 HgI2 + 2KNO3
HgI2 + 2KI K2HgI4
Na2[Fe(CN)5NO] 2Na+ + [Fe(CN)5NO]2-
Hg[Fe(CN)5NO]Hg2+ + [Fe(CN)5NO]2-
Cl
-
+ Hg2+ [HgCl]+
[HgCl]+ + Cl
-
HgCl2
OH2H2O
+ Zn2+ + 2H+
комплекс індикатору з Zn2+ (червоного кольору)вільний індикатор (синього кольору)
OH
NaO3S
O2N
OH
N N
O2N
NaO3S
O Zn O
NN](https://image.slidesharecdn.com/1-170625000218/85/1-82-320.jpg)
![84
Надлишкова крапля титранту реагує з комплексом – комплекс руйнується
і випадає осад меркурію (II) йодиду червоного кольору; таким чином
використання індикатору при титруванні йодидів не потрібно:
Молярна маса еквіваленту в цьому випадку дорівнює двом молекулярним
масам: Е = 2М.м.
Переваги меркуріметричного методу:
1. Титрування галогенідів у кислому середовищі.
2. Багато йонів, які заважають визначенню при використанні методів Мора і
Фольгарда, у даному випадку не впливають на результат аналізу.
3. Не застосовуються дорогі та дефіцитні солі аргентуму.
4. Сполуки меркурію (II) легко регенеруються.
3.2.4. Кислотно-основне титрування у водному середовищі
Згідно з найбільш визнаною теорією кислот і основ – протолітичною
теорією Бренстеда і Лоурі – кислотно-основні реакції здійснюються за рахунок
переносу протону від кислоти до основи. Інакше кажучи, кислота є донором, а
основа – акцептором протонів.
Кислота і основа, які відрізняються вмістом протону, називаються
супряженими, наприклад: H2O і OH-; NH4
+
і NH3; CH3COOH і CH3COO-.
Метод кислотно-основного титрування ґрунтується на реакціях кислотно-
основної взаємодії, які в загальному вигляді можна представити таким чином:
Кислоти (нейтральні молекули, катіони або аніони) можуть віддавати
протон розчиннику або протонкоординуючий основі, при цьому в водних
розчинах утворюється іон гідроксонію H3O+
або в загальному випадку онієвий
іон. Чим дужчою є донорна кислота, тим слабша відповідна акцепторна основа.
Сила кислоти або основи значною мірою залежить від кислотно-основних
властивостей розчинника. В будь-якому розчиннику найдужчою кислотою є
сольватований протон – іон ліолію, а найдужчою основою – іон ліата (аніон
розчинника). Так, в водному розчині найдужчою кислотою є іон гідроксонію
H3O+
, а найдужчою основою – іон гідроксилу OH-; в рідкому амоніаку
найдужча кислота – іон амонію NH4
+
, а найдужча основа – амід-іон NH2
-; в
льодяній оцтовій кислоті найдужча основа – ацетат-іон CH3COO-, а найдужча
кислота – іон ацетонію CH3COOH2
+
.
Розчинення багатьох речовин можна представити як реакцію утворення
супряжених кислоти і основи:
K2[HgI4] + Hg(NO3)2 2HgI2 + 2KNO3
HA + B HB+ + A
-
кислота
кислота
супряжена супряженаоснова
основа](https://image.slidesharecdn.com/1-170625000218/85/1-83-320.jpg)
![85
Сила кислоти в водному розчині визначається тим, наскільки повно вона
віддає протони молекулам води, а сила основи – наскільки повно вона акцептує
протони молекул води.
Реакція між хлороводневою кислотою та водою протікає практично до
кінця, тому хлороводнева кислота належить до класу сильних кислот.
Сильними кислотами в водних розчинах є хлорна, хлороводнева, бромоводнева,
йодоводнева, сульфатна та нітратна кислоти.
Оцтова кислота реагує з водою меншою мірою, тому оцтова кислота
вважається слабкою кислотою.
Сила кислоти або основи оцінюється константою дисоціації, яка є
окремим випадком константи рівноваги, та виражається для кислот рівнянням:
а для основ:
Чим більшою є константа дисоціації, тим більша сила кислоти або
основи.
У випадку водних розчинів реакцію нейтралізації можна представити
таким чином:
При титруванні розчинів сильних кислот сильними основами і сильних
основ сильними кислотами у точці еквівалентності середовище нейтральне.
Після досягнення точки еквівалентності спостерігається різка зміна (стрибок)
pH, яку можна зафіксувати або інструментальними методами (наприклад,
потенціометричним), або візуально за допомогою кислотно-основних
індикаторів.
Кислотно-основні індикатори за своєю природою, як правило, слабкі
органічні кислоти або основи; при відщепленні або приєднанні протону
відбувається зміна їх забарвлення, що дає змогу використовувати ці сполуки
для фіксування кінцевої точки титрування. Найчастіше для аналізу лікарських
речовин застосовують такі кислотно-основні індикатори (табл. 3.1):
[B]
[BH+] [OH-
]
Kb =
H3O+ + OH
-
2H2O
CH3COOH + H2O CH3COO-
+ H3O+
NH3 + H2O NH4
+ + OH
-
HCl + H2O Cl
-
+ H3O+
[HA]
[H3O+] [A-
]
Ka = ,](https://image.slidesharecdn.com/1-170625000218/85/1-84-320.jpg)



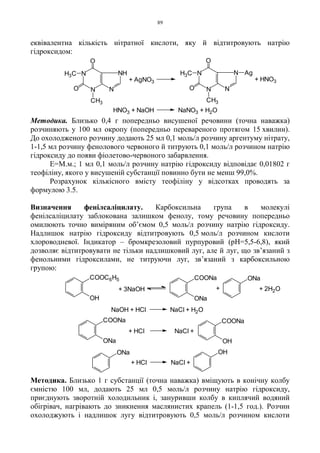



![93
Методика. Близько 1,5 г субстанції (точна наважка) розчиняють у 20 мл води в
колбі з притертою пробкою ємністю 250 мл, додають 45 мл ефіру, 3-4 краплі
змішаного індикатору (1 мл розчину метилового оранжевого і 1 мл розчину
метиленового синього) і титрують 0,5 моль/л розчином кислоти хлороводневої
до появи бузкового забарвлення водного шару. В кінці титрування вміст колби
добре перемішують.
Е=М.м.; 1 мл 0,5 моль/л розчину кислоти хлороводневої відповідає
0,07205 г натрію бензоату, якого в перерахунку на суху речовину повинно бути
не менш 99,0%.
Розрахунок кількісного вмісту натрію бензоату у відсотках проводять за
формулою 3.9.
Непряме ацидиметричне титрування (за замісником)
Визначення меркурію окису жовтого. При взаємодії меркурію(ІІ) оксиду з
розчином калію йодиду утворюється розчинна комплексна сіль та луг, який
відтитровують кислотою:
HgО + 4KI + H2O → K2[HgI4] + 2KOH
2KOH + 2HCl → 2KCl + 2H2O
Сумарно: HgO + 4KI + 2HCl → K2[HgI4] + 2KCl + H2O
Методика. Близько 0,2 г субстанції (точна наважка) збовтують з 5 мл води,
додають 2 г калію йодиду та знову збовтують до повного розчинення. Після
цього додають ще 20 мл води і титрують 0,1 моль/л розчином кислоти
хлороводневої до появи рожевого забарвлення (індикатор – метиловий
червоний).
Е=1/2М.м.; 1 мл 0,1 моль/л розчину кислоти хлороводневої відповідає
0,01083 г меркурію окису, кількісний вміст якого повинен бути не менш 99,0%.
Розрахунок кількісного вмісту меркурію окису жовтого ведуть за
формулою 3.5.
3.2.5. Кислотно-основне титрування в неводному середовищі
Метод кислотно-основного титрування в неводному середовищі
застосовується для кількісного визначення лікарських речовин, які є слабкими
основами або кислотами (Кдис.<1.10-8
), їх солей, а також речовин, які погано
розчиняються у воді.
Під впливом різних розчинників властивості однієї і тієї ж речовини
можуть різко змінюватися. Сила кислоти або основи визначається ступенем їх
взаємодії з розчинником. Правильно підібраний неводний розчинник може
посилювати основні або кислотні властивості слабкої основи або слабкої
+ NaCl+ HCl
COONa COOH](https://image.slidesharecdn.com/1-170625000218/85/1-92-320.jpg)




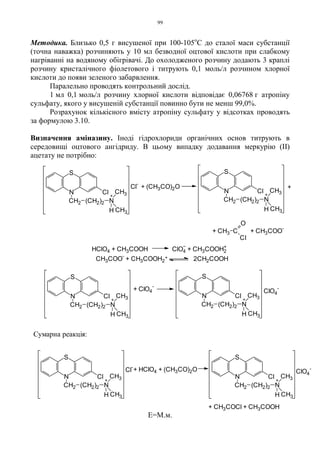



![103
Визначення натрію нітриту. Натрію нітрит в кислому середовищі
розкладається з виділенням нітрозних газів, тому метод прямого титрування
застосовувати не можна. В кількісному визначенні натрію нітриту
використовують метод зворотного титрування з йодометричним визначенням
надлишку калію перманганату:
2KMnO4 + 5NaNO2 + 3H2SO4 → K2SO4 + 2MnSO4 + 5NaNO3 + 3H2O
2KMnO4 + 10KI + 8H2SO4 → 6K2SO4 + 5I2 + 2MnSO4 + 8H2O
I2 + 2Na2S2O3 → 2NaI + Na2S4O6
Методика. Близько 1 г субстанції (точна наважка) розчиняють в мірній колбі
ємністю 100 мл і розчин доводять водою до мітки. Точно відмірюють 10 мл
цього розчину і повільно виливають в суміш з 40 мл 0,1 моль/л розчину калію
перманганату, 300 мл води та 25 мл розведеної сульфатної кислоти. Через 20 хв
до одержаної рідини додають 2 г калію йодиду і йод, що виділився, титрують
0,1 моль/л розчином натрію тіосульфату (індикатор – крохмаль).
Паралельно проводять контрольний дослід.
Е=1/2М.м.; 1 мл 0,1 моль/л розчину натрію тіосульфату відповідає
0,003450 г натрію нітриту, якого в субстанції повинно бути не менш 98%.
Розрахунок кількісного вмісту натрію нітриту у відсотках проводять за
формулою:
п
мкк
Vm
VTKVV
X
⋅
⋅⋅⋅⋅−
=
100)(
% , (3.13)
де Vк – об’єм 0,1 моль/л розчину натрію тіосульфату, витраченого на
титрування у контрольному досліді, мл;
V – об’єм 0,1 моль/л розчину натрію тіосульфату, витраченого на
титрування в прямому досліді, мл;
Vмк – об’єм мірної колби, мл;
Vп – об’єм піпетки, мл;
К –коефіцієнт поправки до молярності 0,1 моль/л розчину
натрію тіосульфату;
Т – титр 0,1 моль/л розчину натрію тіосульфату за натрію нітритом, г/мл;
m – маса наважки натрію нітриту, г.
3.2.6.2. Йодометрія
Йодометрія – метод кількісного визначення вільного йоду, тих речовин,
які кількісно виділяють його під час реакцій і тих сполук, які зв’язують йод або
окиснюються йодом в стехіометричних кількостях.
Йодометричний метод кількісного визначення має широке практичне
застосування; за своєю простотою і точністю він визнається одним з кращих
редокс-методів кількісного визначення.
В основі йодометричного визначення лежать реакції:
[I3] + 2e 3I
3I
_
2e [I3]](https://image.slidesharecdn.com/1-170625000218/85/1-102-320.jpg)
![104
Нормальний окисно-відновний потенціал цієї системи дорівнює 0,545 В.
Ті речовини, які мають більш низький потенціал окиснюються йодом, а
речовини, що мають більш високий потенціал, окиснюють йодид-іони до йоду,
котрий потім може бути відтитрований за реакцією:
2S2O3
2-
+ [I3]- → S4O6
2-
+ 3I-
Нормальний окисно-відновний потенціал системи S4O6
2-
/2S2O3
2-
дорівнює
0,17 В.
Пряме йодометричне титрування. Методом прямого йодометричного
титрування визначають речовини, які мають сильні відновні властивості
(натрію тіосульфат, аскорбінова кислота, лікарські сполуки арсену (III) та інші).
Визначення проводять в кислому, нейтральному або слабко-лужному
середовищі. Титрантом є розчин йоду в йодиді калію. Цей розчин має жовто-
бурий колір і зайва його крапля забарвлює розчин, що титрується, в блідо-
жовтий колір, що може слугувати ознакою кінця титрування (кількісне
визначення анальгіну). Іноді рекомендують додавати декілька мілілітрів
органічного розчинника, що не змішується з водою, наприклад, хлороформу.
При збовтуванні надлишковий йод переходить в хлороформний шар і надає
йому фіолетового забарвлення.
Однак найбільш чітко кінцеву точку титрування можна визначити за
допомогою крохмалю, який з йодом в присутності йодид-іонів утворює
комплексну сполуку інтенсивно-синього кольору.
Зворотна йодометрія. Методом зворотної йодометрії визначають сполуки, які
повільно окиснюються йодом (ізоніазид), які утворюють з ним комплексні
сполуки (кофеїн), дають реакцію ароматичного заміщення (антипірин) або
потребують для стехіометричного необоротного окиснення лужного
середовища (формальдегід, глюкоза, фурацилін). В останньому випадку
окиснення відбувається за схемою:
Після завершення реакцій надлишок йоду відтитровують тіосульфатом
натрію. Якщо окиснення проводили в лужному середовищі, до реакційної
суміші спочатку додають надлишок кислоти, а тоді йод, що виділився,
відтитровують тіосульфатом натрію:
При проведенні зворотного йодометричного визначення крохмаль
додають в кінці титрування, коли розчин набуде блідо-жовтого кольору –
з’являється інтенсивне синє забарвлення – і далі титрують до знебарвлення.
Додавати крохмаль до розчинів з великою концентрацією йоду не можна,
оскільки в цьому випадку відбувається необоротне зв’язування йоду.
[I3] + 2OH 2I + IO + H2O
IO + H2O + 2e I + 2OH
[I3]IO + 2I + 2H+ + H2O](https://image.slidesharecdn.com/1-170625000218/85/1-103-320.jpg)





![110
Визначення кислоти аскорбінової. Визначення ґрунтується на відновних
властивостях кислоти аскорбінової. Титрантом-окисником є калію йодат в
присутності калію йодиду:
Е=1/2М.м.
Надлишок титрованого розчину калію йодату в кислому середовищі
окиснює калію йодид:
KIO3 + 5KI + 6HCl → 3I2 + 6KCl + 3H2O
Йод, що виділився, забарвлює крохмаль в синій колір.
Методика. Близько 0,5 г субстанції (точна наважка) розчиняють у воді в мірній
колбі ємністю 50 мл, доводять об’єм розчину водою до мітки та перемішують.
До 10 мл приготованого розчину додають 0,5 мл 1% розчину калію йодиду,
2 мл розчину крохмалю та 1 мл 2% розчину хлороводневої кислоти і титрують
0,1 моль/л розчином калію йодату до появи стійкого синього забарвлення.
1 мл 0,1 моль/л розчину калію йодату відповідає 0,008806 г кислоти
аскорбінової, якої в субстанції повинно бути не менш 99,0%.
Розрахунок кількісного вмісту кислоти аскорбінової у відсотках
проводять за формулою 3.7.
3.2.6.5. Нітритометрія
Нітритометрія – титриметричний метод аналізу, який ґрунтується на
окисно-відновних властивостях системи HNO2/NO, Eo
=0,99 B. Редокс-потенціал
системи достатньо великий, тому нітритометрично можна визначати цілий ряд
відновників (As2O3, FeSO4).
Але найчастіше нітритометрію застосовують для кількісного визначення
органічних лікарських речовин, які мають у своєму складі первинну чи
вторинну ароматичні аміногрупи, або нітрогрупу, яку перед визначенням
відновлюють до аміногрупи. Як титрант застосовують натрію нітрит, з якого у
кислому середовищі виділяється нітритна кислота. У загальному вигляді
реакцію діазотування можна представити таким чином:
З наведеного рівняння видно, що в ньому беруть участь 2 молекули
кислоти, з яких одна йде на утворення нітритної кислоти, а інша – солі
діазонію. Однак, для того, щоб реакція проходила стехіометрично, необхідна
+
ArNH2 + 2HCl + NaNO2 [Ar N N] Cl
-
+ NaCl + 2H2O
O
O
OHOH
H
CH
CH2OH
HO
3 + KIO3
3
O
O
H
CH
CH2OH
HO
O O
+ KI + 3H2O](https://image.slidesharecdn.com/1-170625000218/85/1-109-320.jpg)

![112
Визначення новокаїну. В основі визначення лежить реакція:
Е=М.м.
Методика. Близько 0,3 г субстанції (точна наважка) розчиняють в суміші 10 мл
води та 10 мл розведеної хлороводневої кислоти і додають воду до загального
об’єму 80 мл. До одержаного розчину додають 1 г броміду калію і при
постійному перемішуванні титрують 0,1 моль/л розчином нітриту натрію,
додаючи його спочатку зі швидкістю 2 мл за хвилину, а в кінці титрування (за
0,5 мл до еквівалентної кількості) по 0,05 мл кожну хвилину. Точку
еквівалентності визначають за допомогою внутрішнього індикатору
тропеоліну-00 в суміші з метиленовим синім. Титрування проводять при
температурі не вище 18-20о
С. Перехід забарвлення від червоно-фіолетового до
блакитного.
1 мл 0,1 моль/л розчину нітриту натрію відповідає 0,02728 г новокаїну,
якого в субстанції повинно бути не менш 99,5%.
Розрахунок кількісного вмісту новокаїну у відсотках проводять за
формулою 3.10.
Визначення парацетамолу. Для того, щоб нітритометрично визначити
парацетамол спочатку проводять його кислотний гідроліз, а потім п-
амінофенол, що утворився, титрують нітритом натрію:
Е=М.м.
Методика. Близько 0,25 г субстанції (точна наважка) вміщують в конічну
колбу ємністю 100 мл, додають 10 мл розведеної хлороводневої кислоти та
кип’ятять зі зворотним холодильником протягом 1 години. Після цього
Cl
-
+ NaCl + 2H2O
. HCl. HCl
+ NaNO2 + 2HCl
+
NH2
COOCH2CH2N
C2H5
C2H5
N N
COOCH2CH2N
C2H5
C2H5
+ CH3COOH+ H2O
[H+]
OH
NHCOCH3
OH
NH2
Cl
-
+ NaCl + 2H2O
+
+ NaNO2 + 2HCl
NH2
OH
N N
OH](https://image.slidesharecdn.com/1-170625000218/85/1-111-320.jpg)









































































