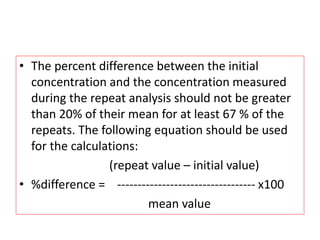
This document provides guidelines for validating bioanalytical methods used to measure drug concentrations in biological matrices. It defines key validation elements including selectivity, calibration curves, accuracy, precision, matrix effects, and stability testing. Guidelines are given for full validation of new methods as well as partial validation when modifying existing methods. Validation ensures reliable results for making critical decisions in drug development.