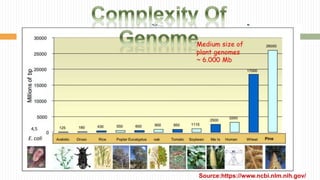
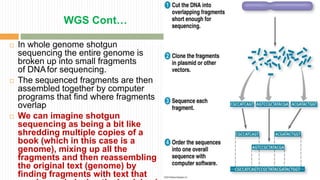
The document discusses the concept of genome and genome sequencing, outlining various methods including first, second, and third generation sequencing techniques. It details case studies on the genome sequencing of crops like rice and pigeon pea, highlighting the challenges and advancements in the field as well as the importance of understanding both coding and non-coding DNA. Lastly, it addresses limitations in current practices and ongoing developments towards more efficient sequencing methodologies for agricultural and scientific applications.