Downloaded 37 times











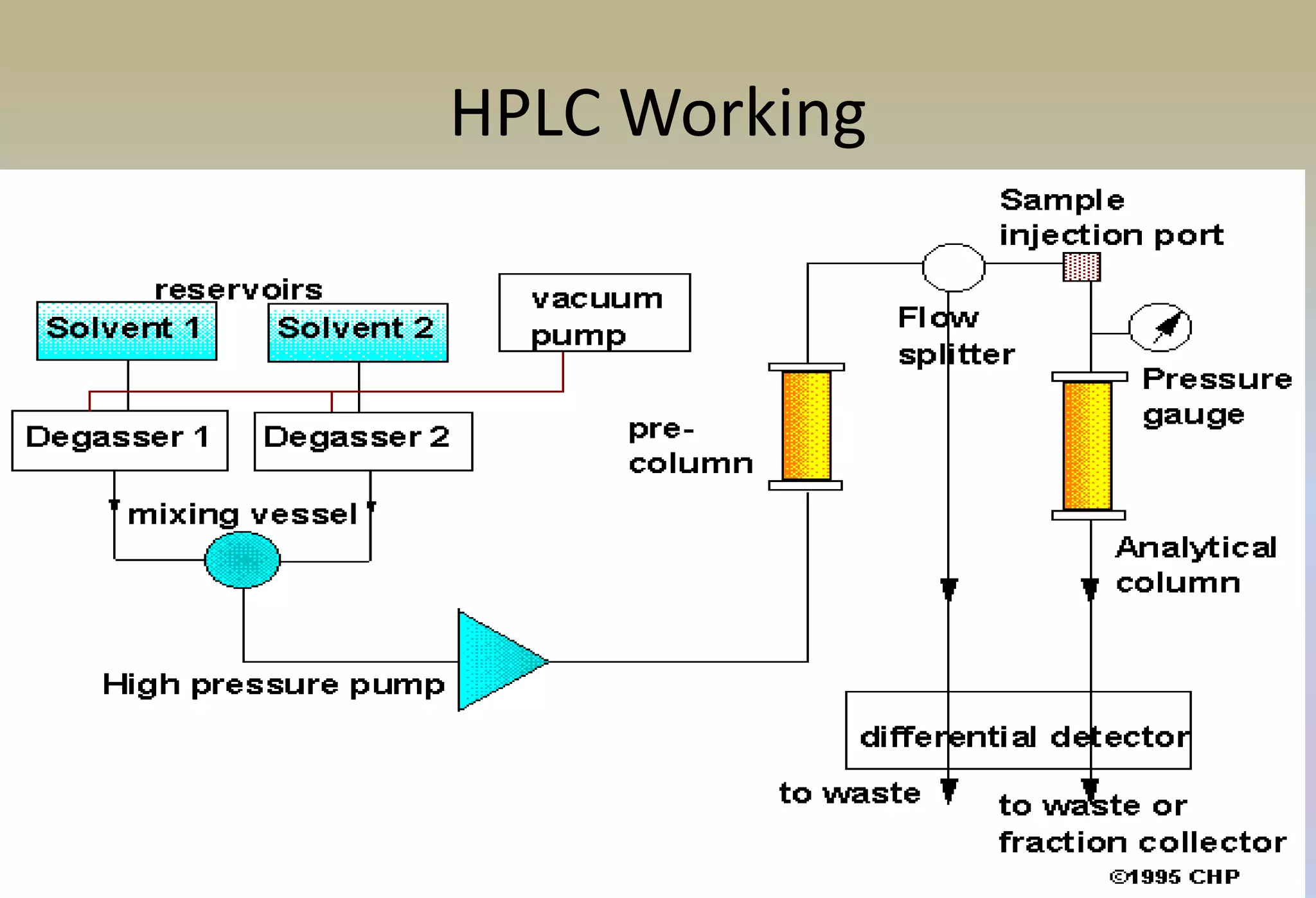
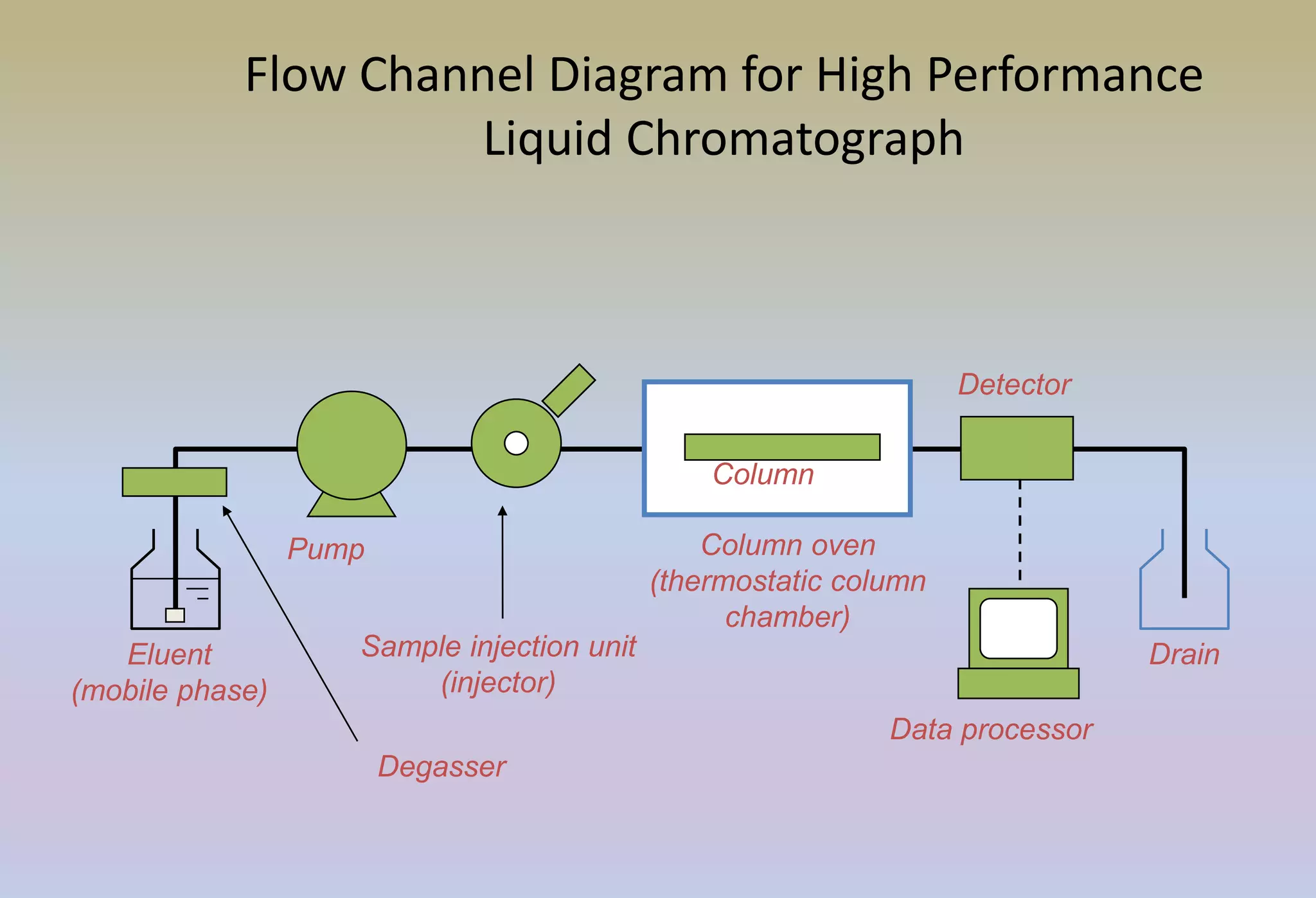










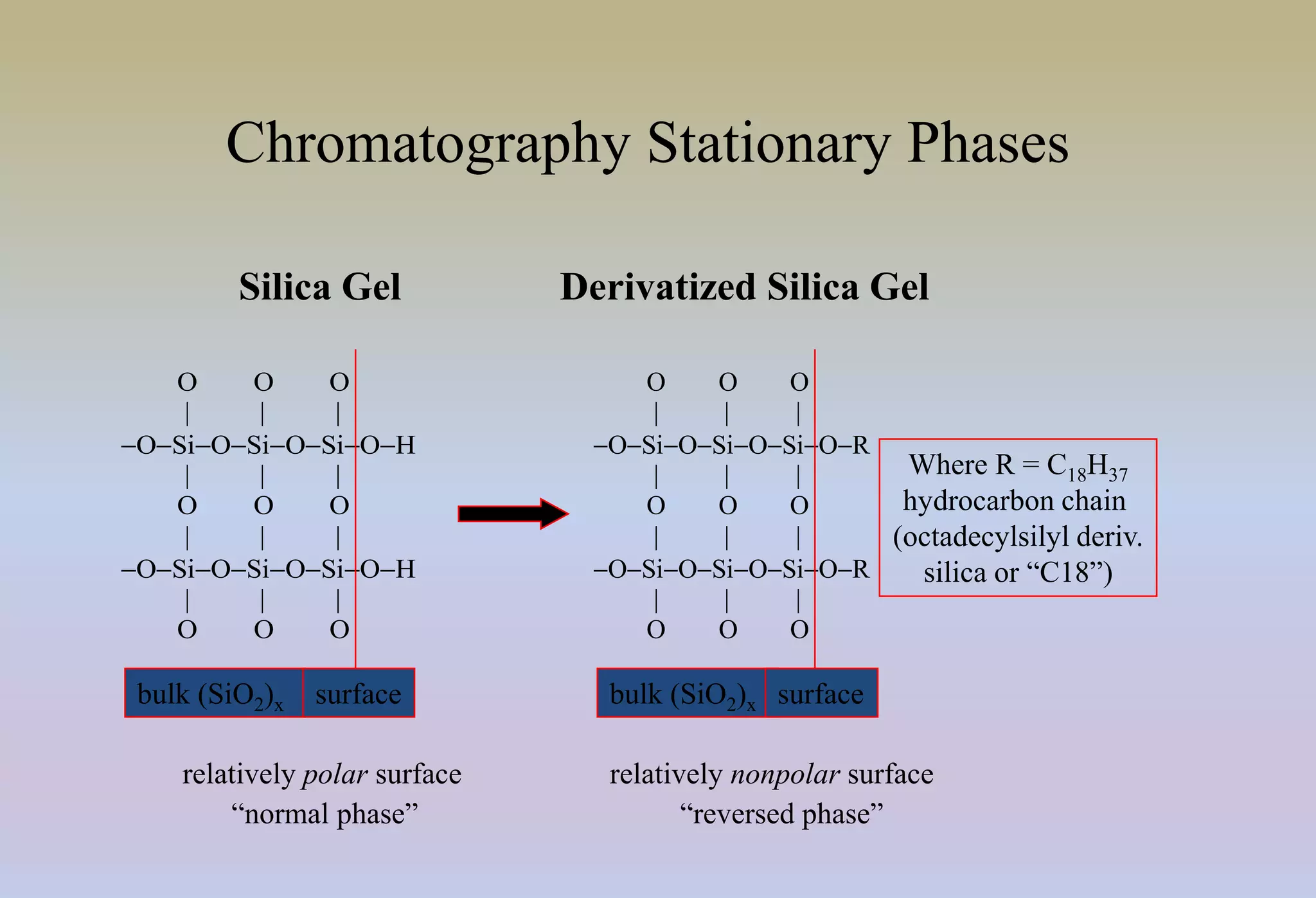













HPLC is characterized by using high pressure to push a mobile phase through a stationary phase column, allowing for the separation of complex mixtures. Validation of analytical methods demonstrates that the methods are accurate, precise, specific, sensitive, linear, and robust for their intended purpose. Key aspects of validation include accuracy, precision, specificity, detection limit, quantitation limit, linearity, range, and robustness. Validation ensures methods provide reliable results.






















































































