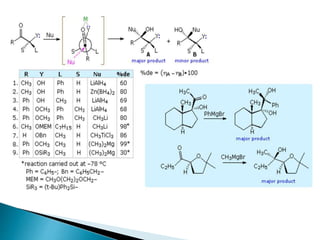
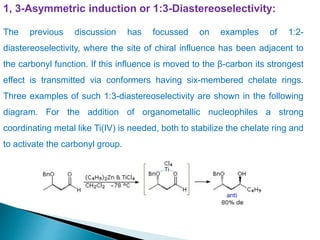
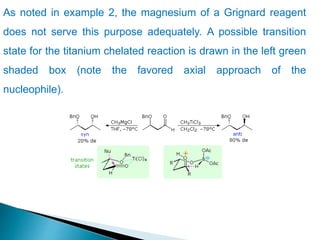
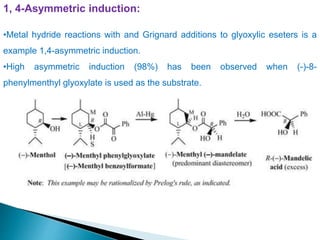
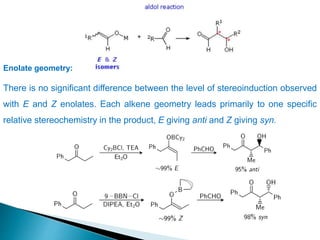
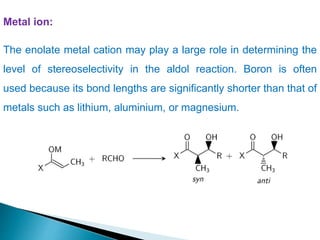
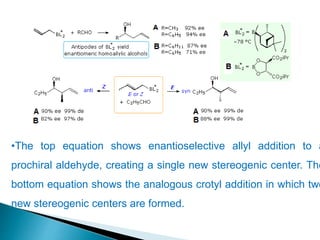
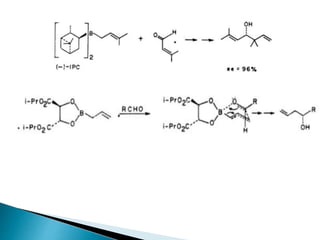
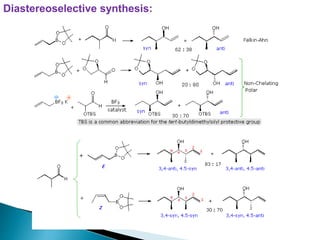
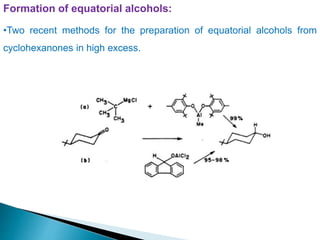
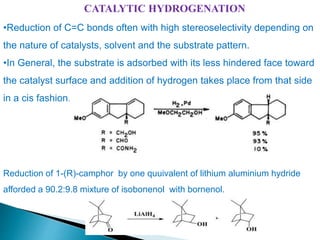
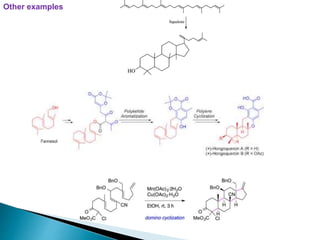
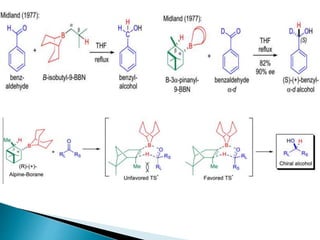
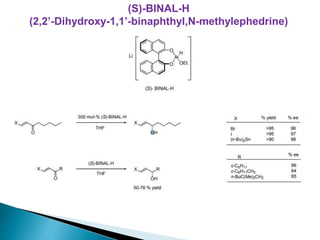
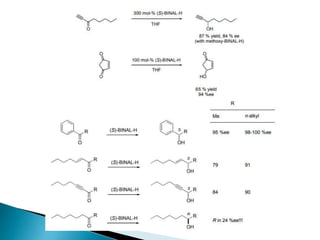
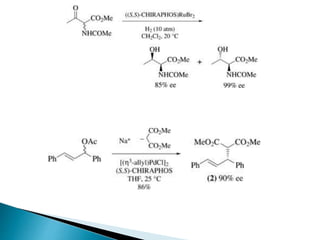
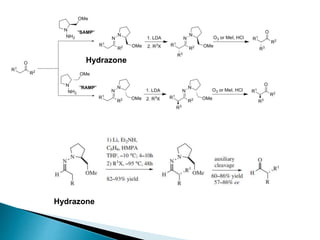
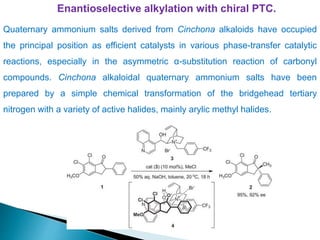
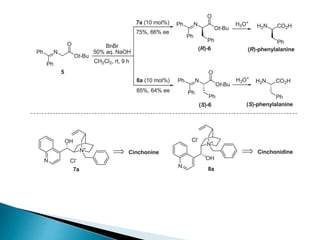
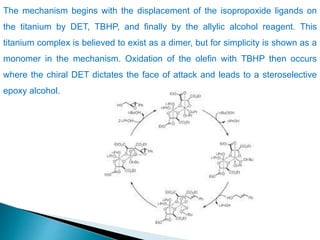
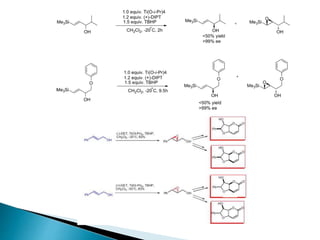
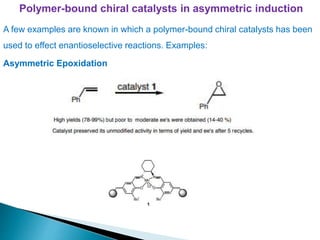
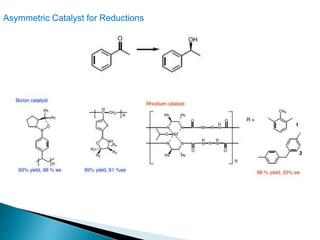
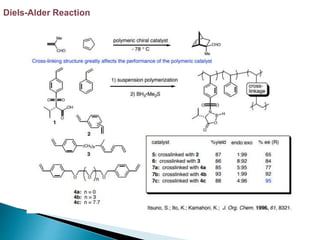
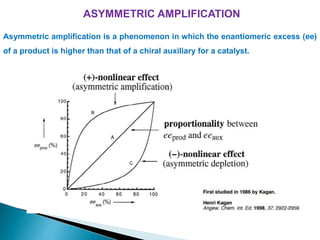
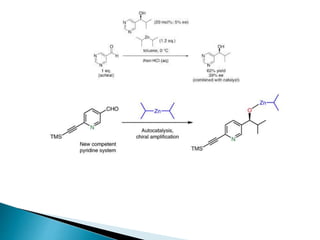
This document discusses various aspects of asymmetric synthesis, including stereochemical aspects, acyclic and cyclic stereoselection, and enantioselective synthesis. It defines terms like racemate, enantiopure, and enantiomer. It describes stereospecific and stereoselective reactions, and rules like Cram's rule and Prelog's rule that help explain stereoselection. It discusses strategies for stereoselective synthesis including additions to carbonyls and aldol reactions. It also covers topics like diastereoselective oxidations, catalytic hydrogenation, and enantioselective reductions using chiral reagents like (S)-PBMgCl and (R,R)-DIOP.






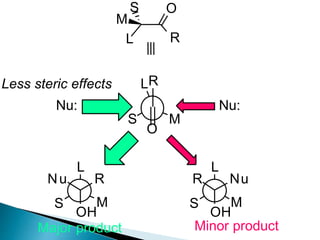
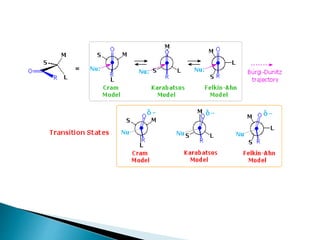
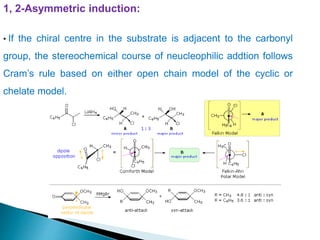
![Cram's rule:
Cram's rule, proposed by Donald Cram in 1952,[1] was the earliest model
proposed for rationalizing the stereoselectivity of nucleophilic additions to
carbonyl groups with α-stereocentres. The model involved assigning each α-
substituent a relative "size" (small, medium, and large), then placing the
carbonyl oxygen antiperiplanar to the largest of these three groups. The
nucleophile then attacks the carbonyl group opposite the larger of the two
remaining groups (i.e., the medium group). This is best explained with a
diagram:
C X
* diastereomeric
faces
X = C, O, N
stereogenic](https://image.slidesharecdn.com/asymmetricsynthesis-ii-200926065553/85/Asymmetric-synthesis-ii-6-320.jpg)











![(S)-PBMgCl
•S-2-Phenyl butyl magnesium chloride [(S)-PBMgCl] is the most
enantioselective.
•The highest enantioselective (82%) has been reported for the reduction of
isopropyl phenyl ketone.
C2H5
MgCl
H
O
(S)-PBMgCl
OH
ee 82%](https://image.slidesharecdn.com/asymmetricsynthesis-ii-200926065553/85/Asymmetric-synthesis-ii-34-320.jpg)
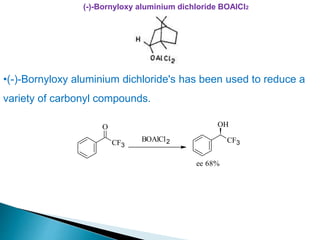
![Alpine-borane or IPC-BBN
• B-(3α-Pinanyl)-9-borabicyclo[3.3.1]nonane.
•Prepared from 1,5-cycloctadiene, borane and α -pinene.
•It is an extremely efficient enantioselective reducing agent.
O
Alphine-borane
OH
ee 83%](https://image.slidesharecdn.com/asymmetricsynthesis-ii-200926065553/85/Asymmetric-synthesis-ii-36-320.jpg)
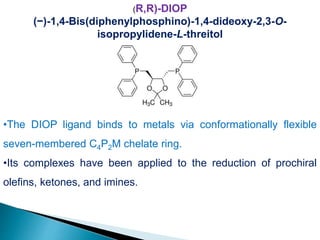
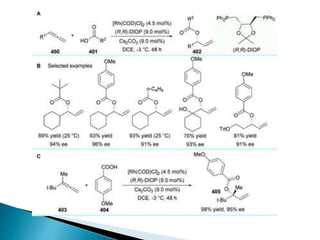
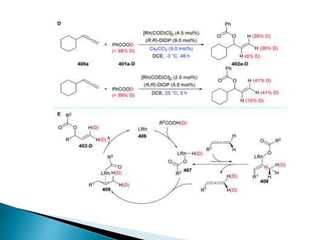
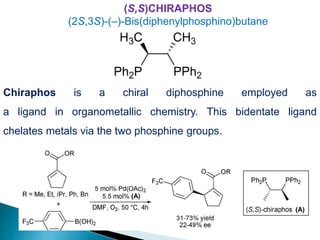






























![Photoisomerisation of aromaric compounds [recovered]](https://cdn.slidesharecdn.com/ss_thumbnails/photoisomerisationofaromariccompoundsrecovered-210612142707-thumbnail.jpg?width=640&height=640&fit=bounds)

























































