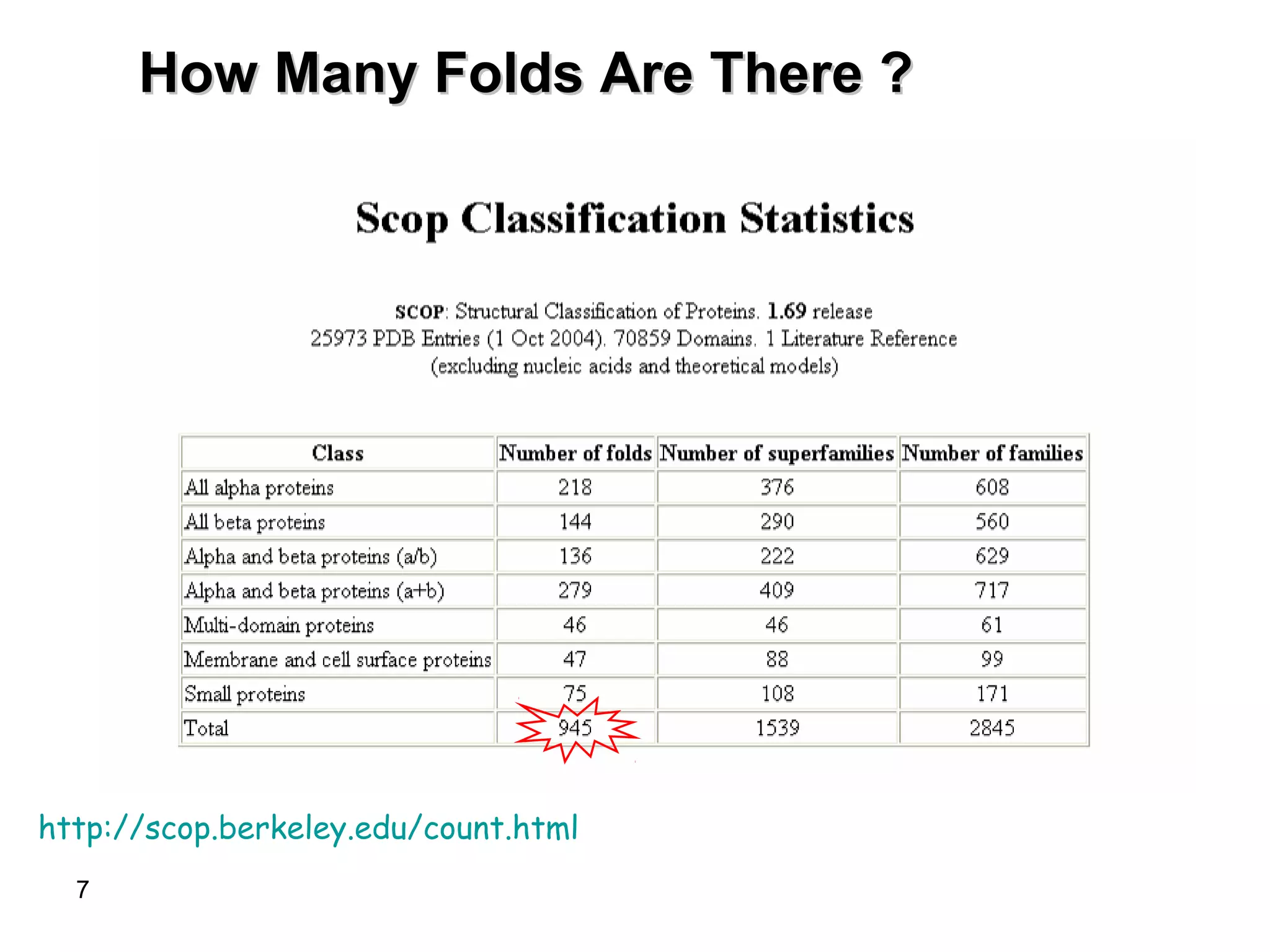
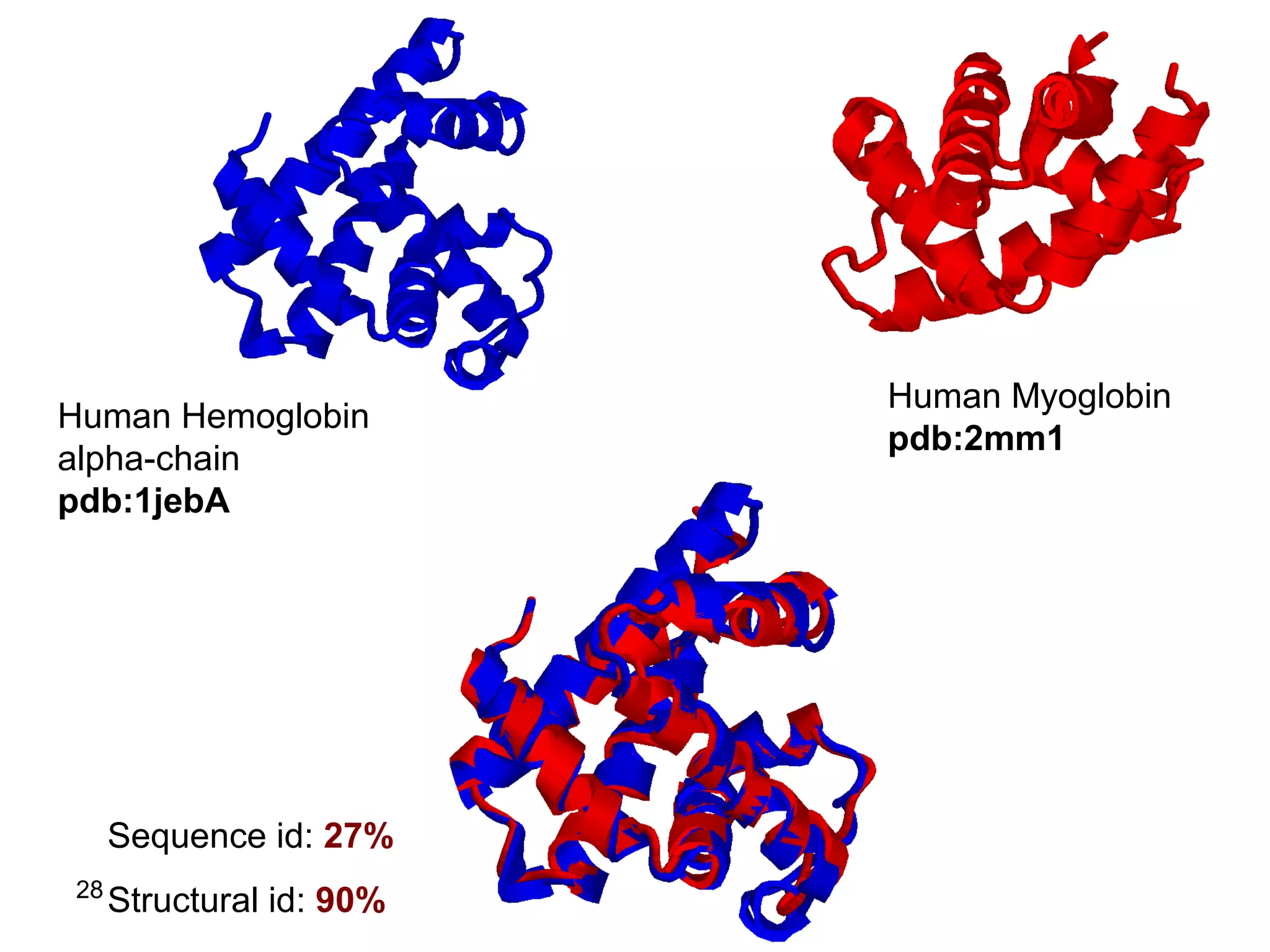
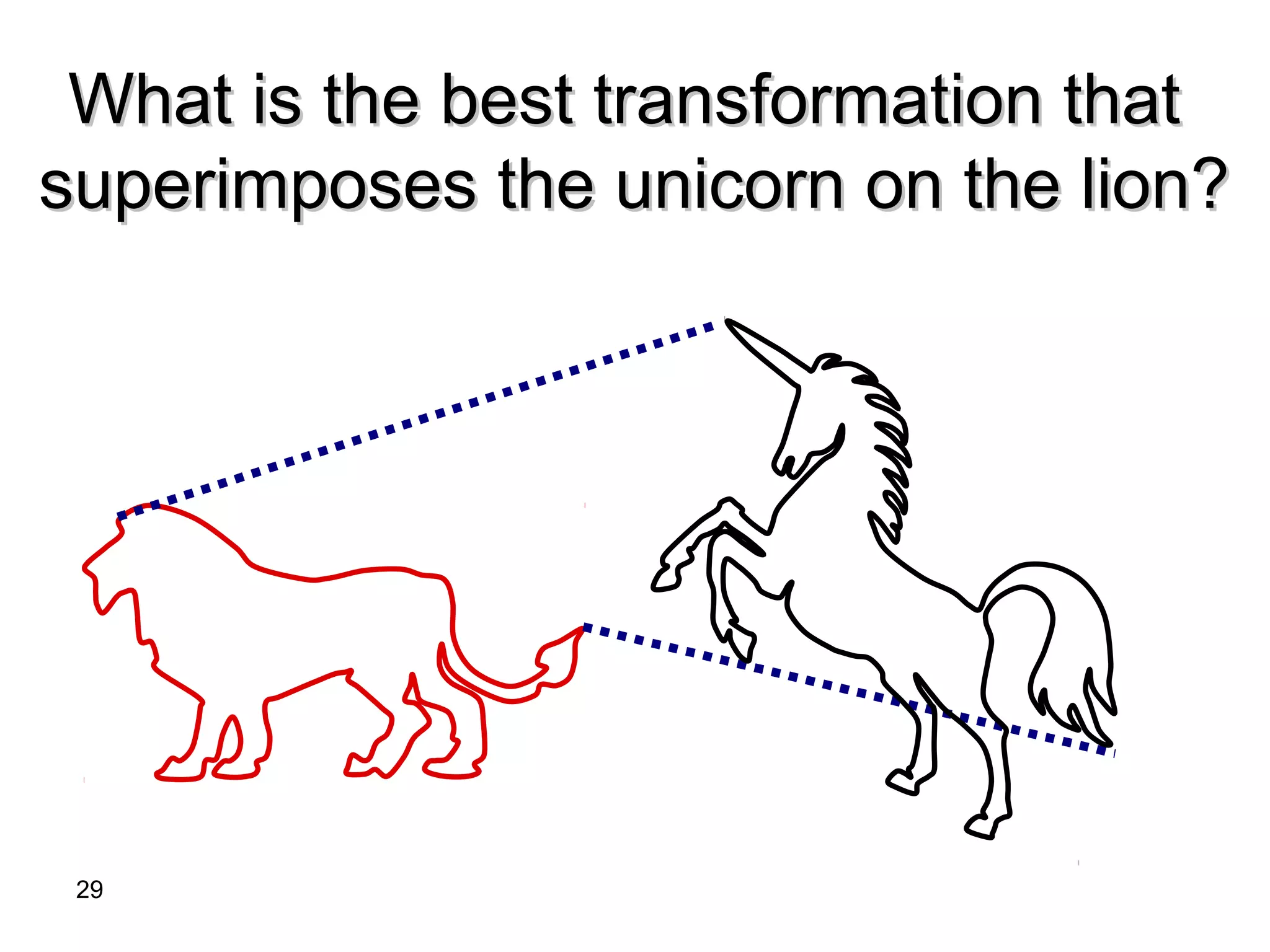
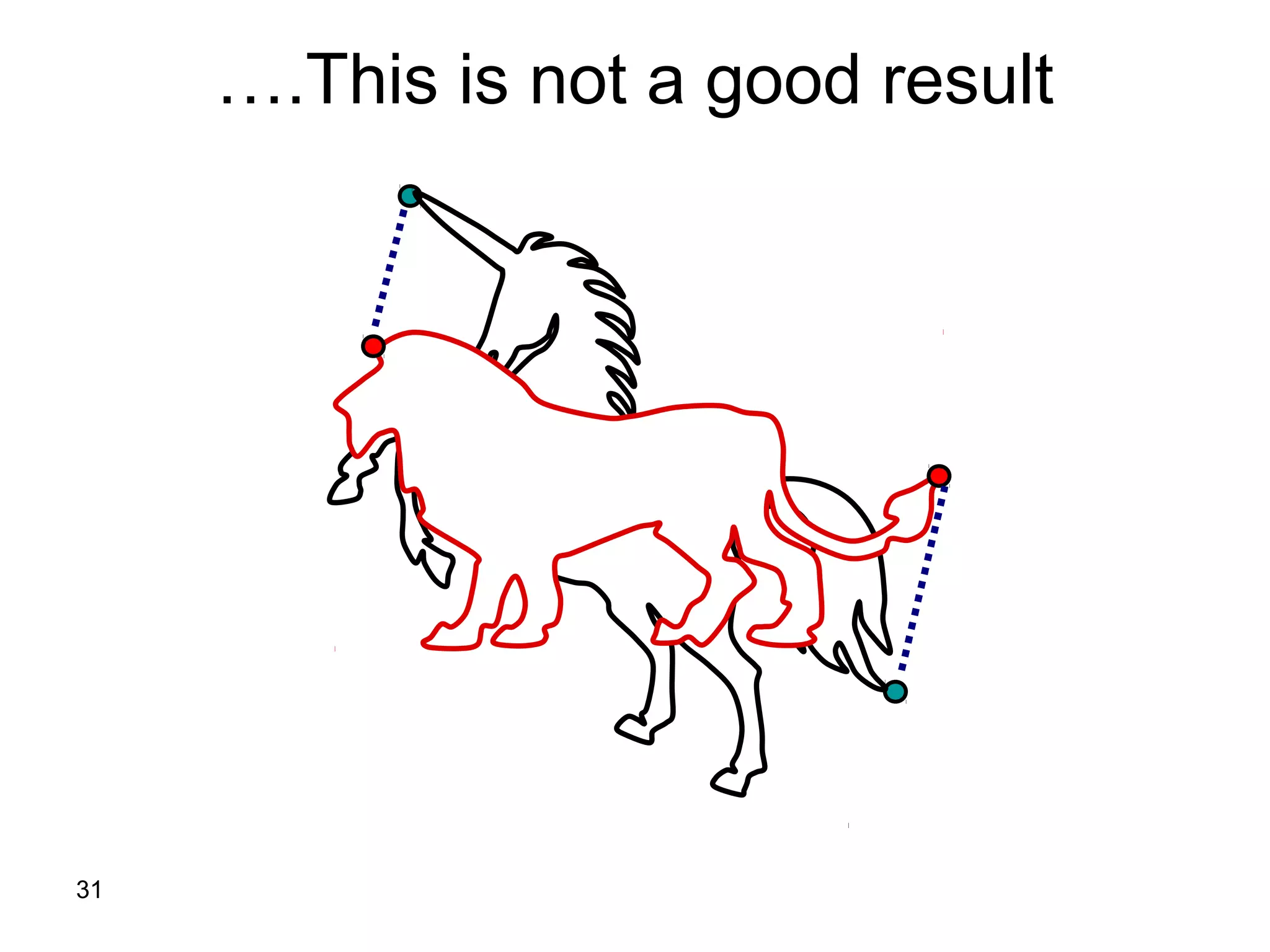
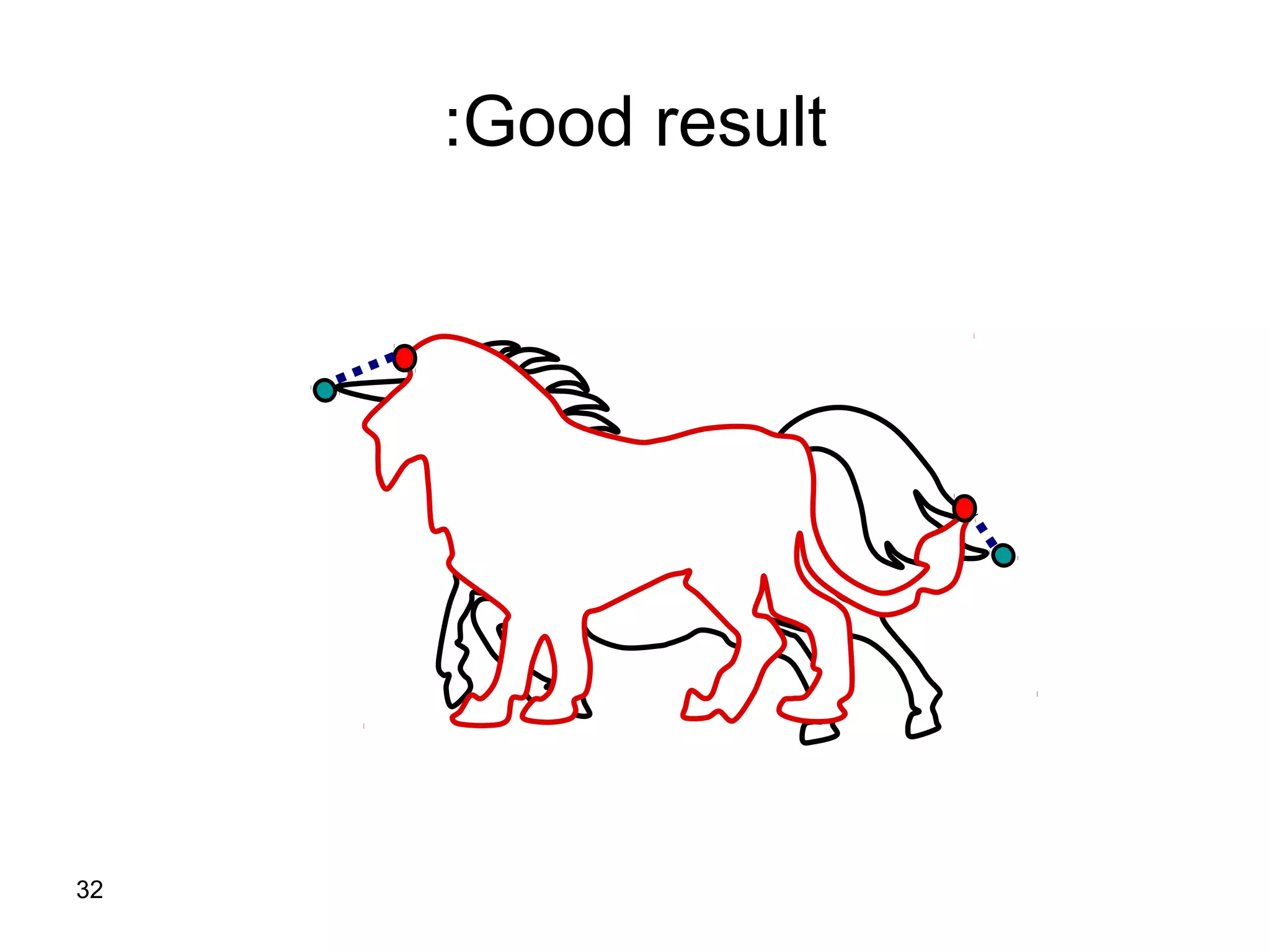
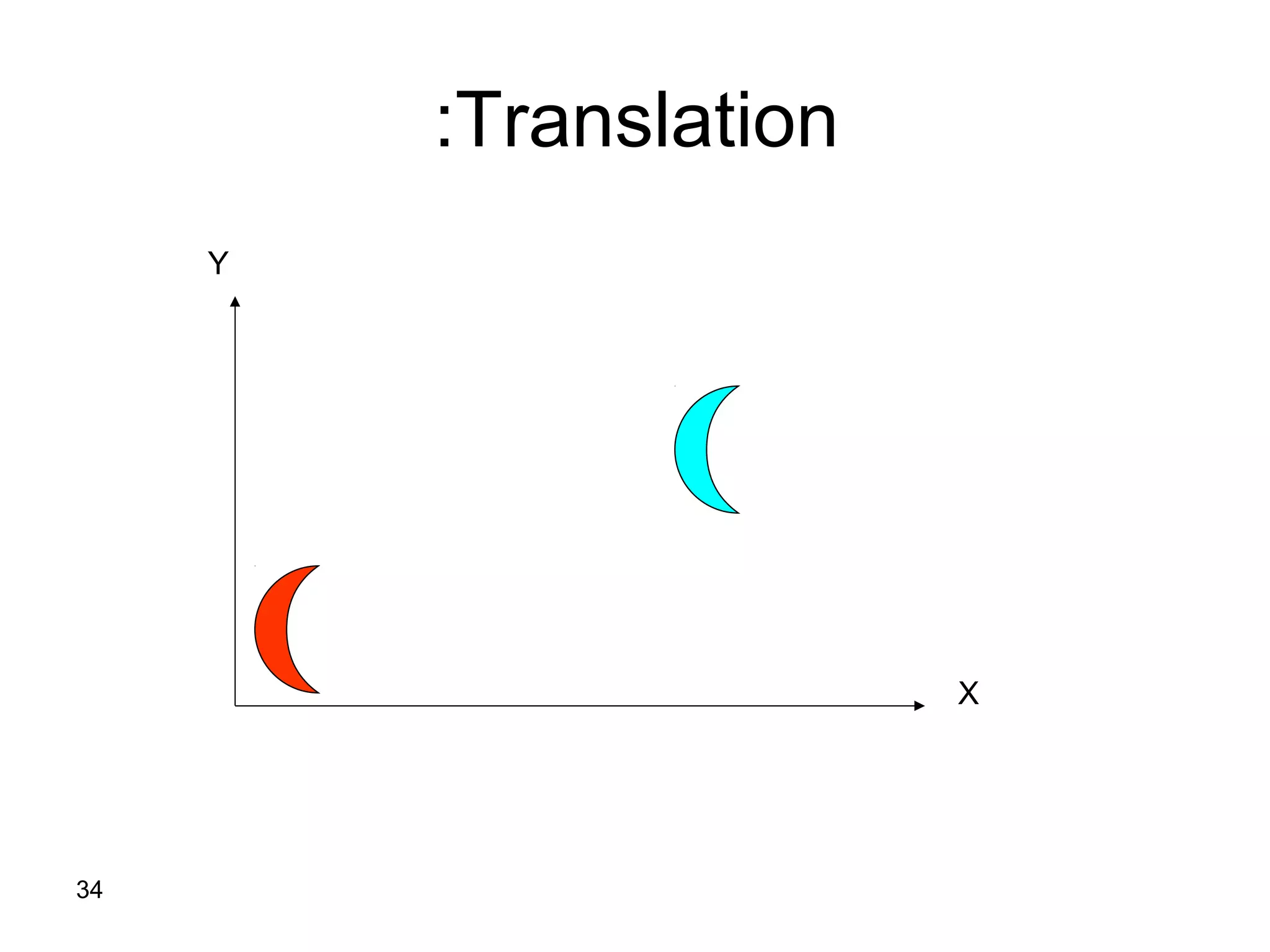
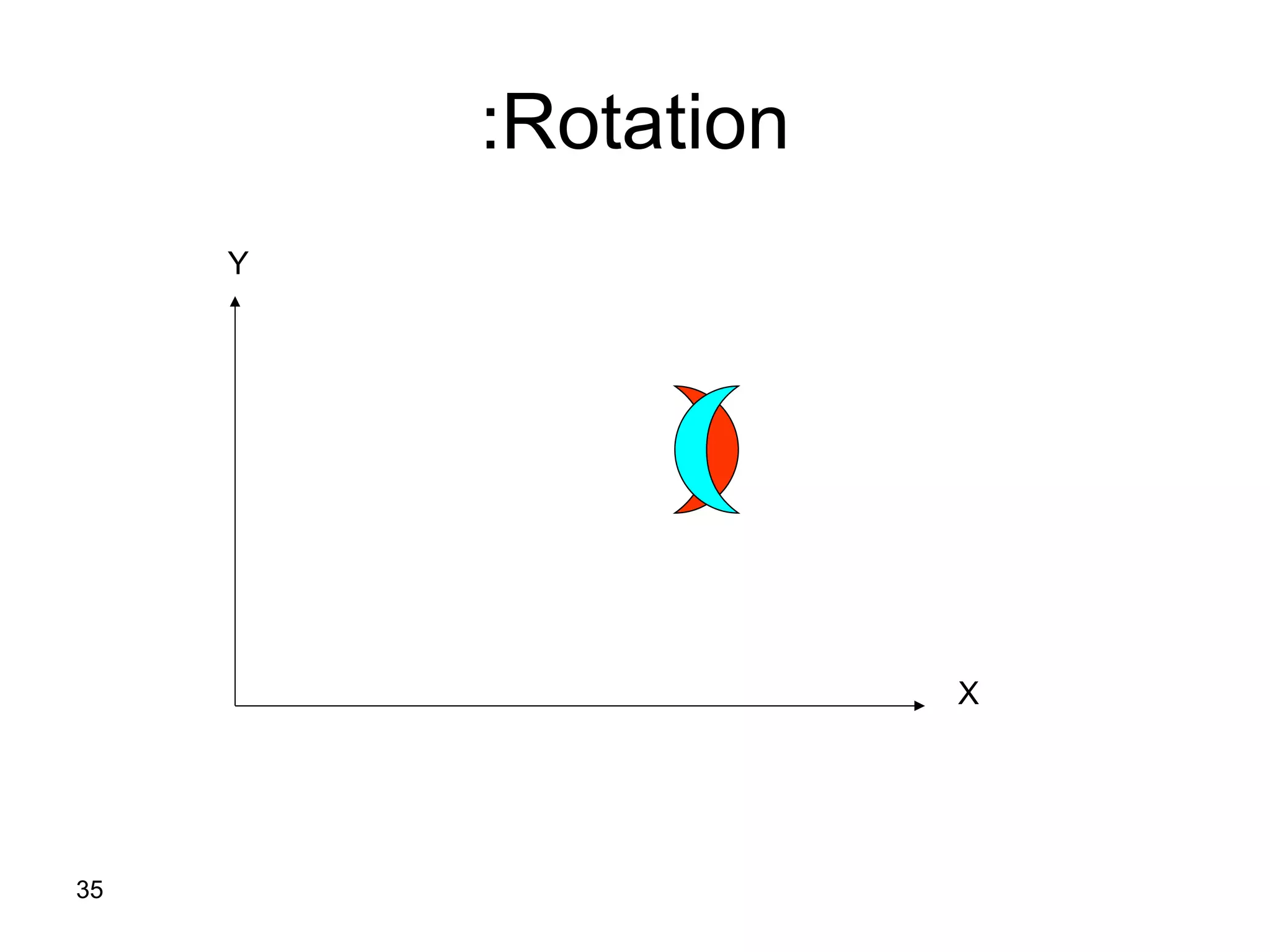
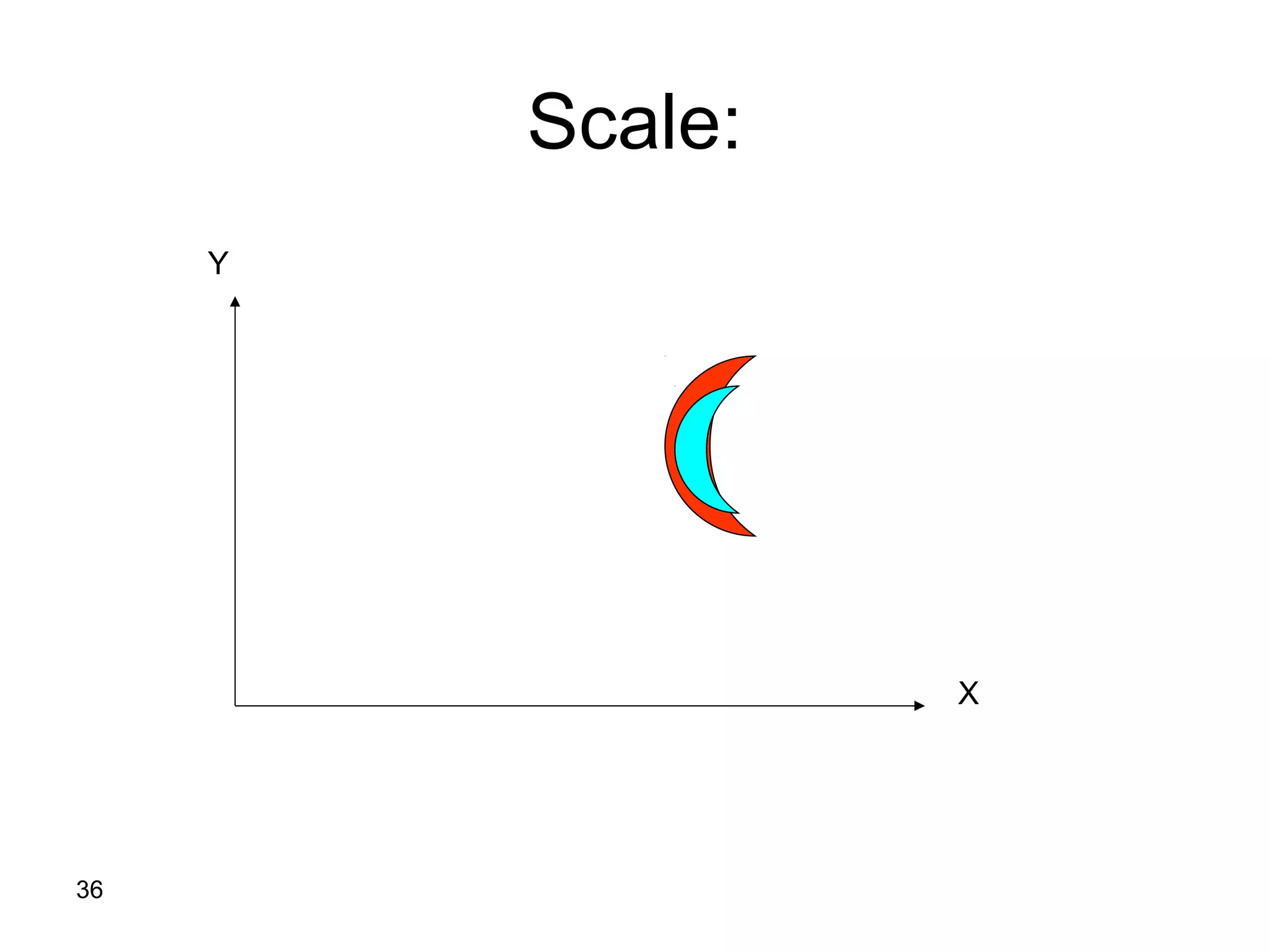
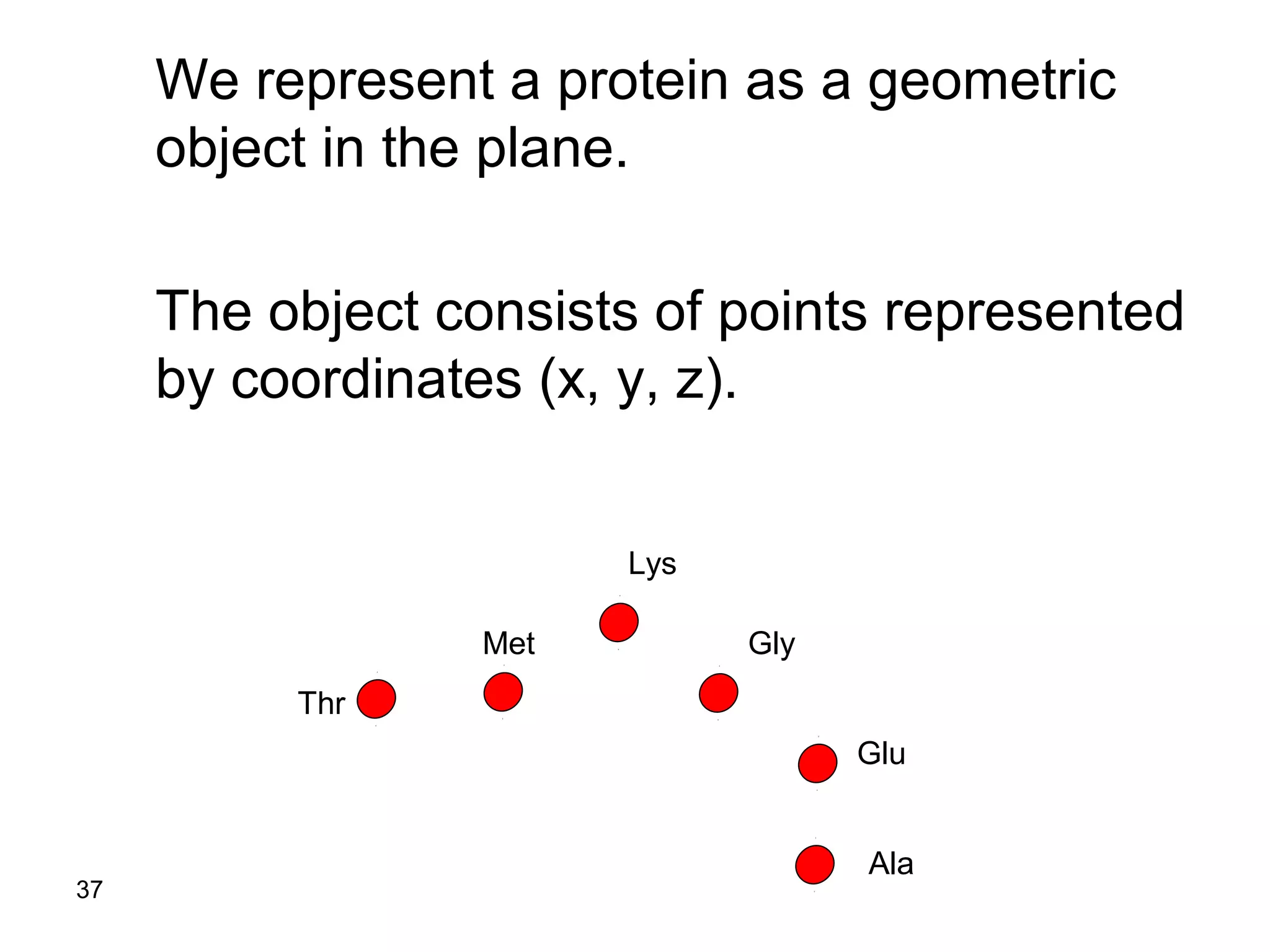
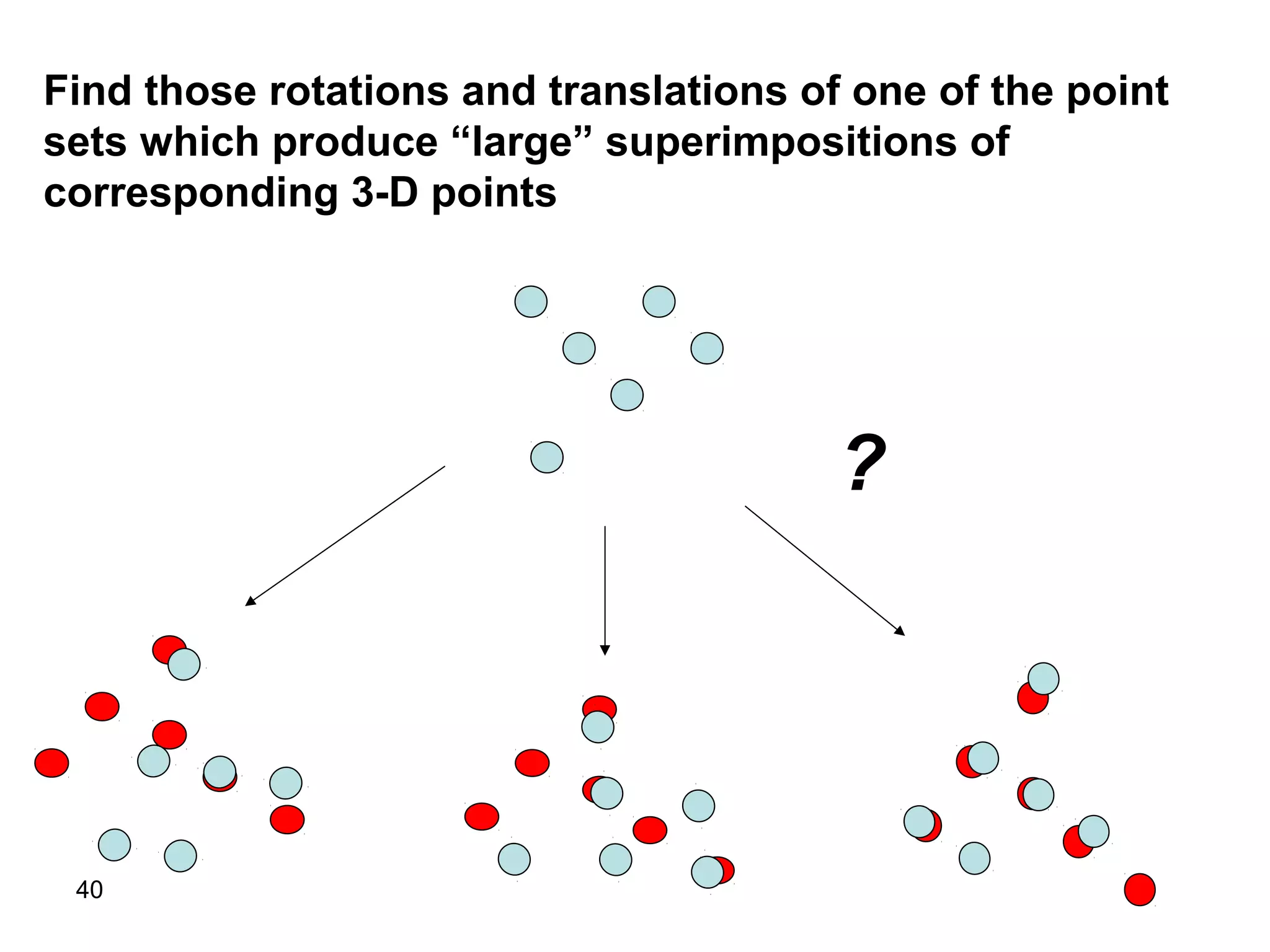
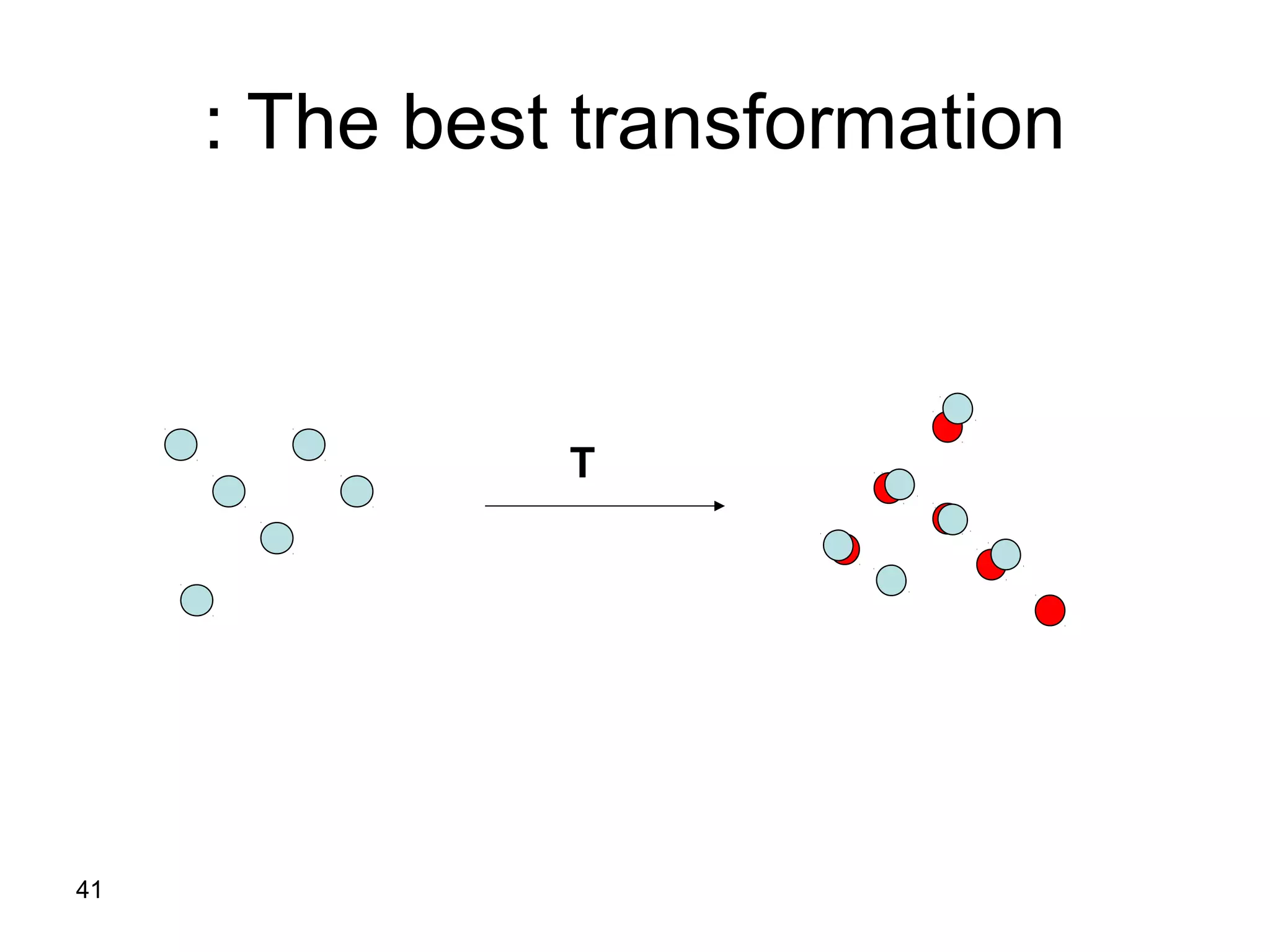
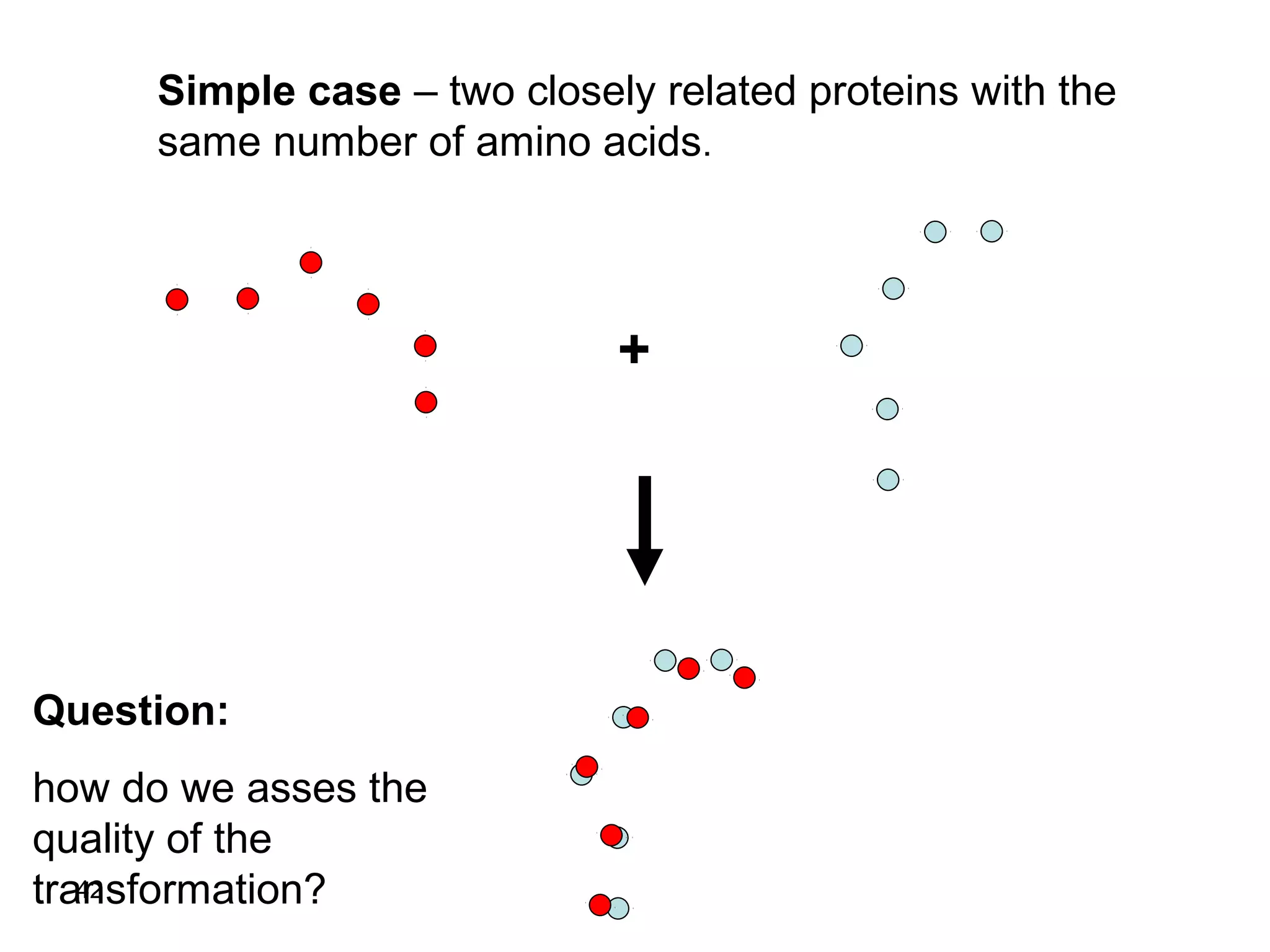
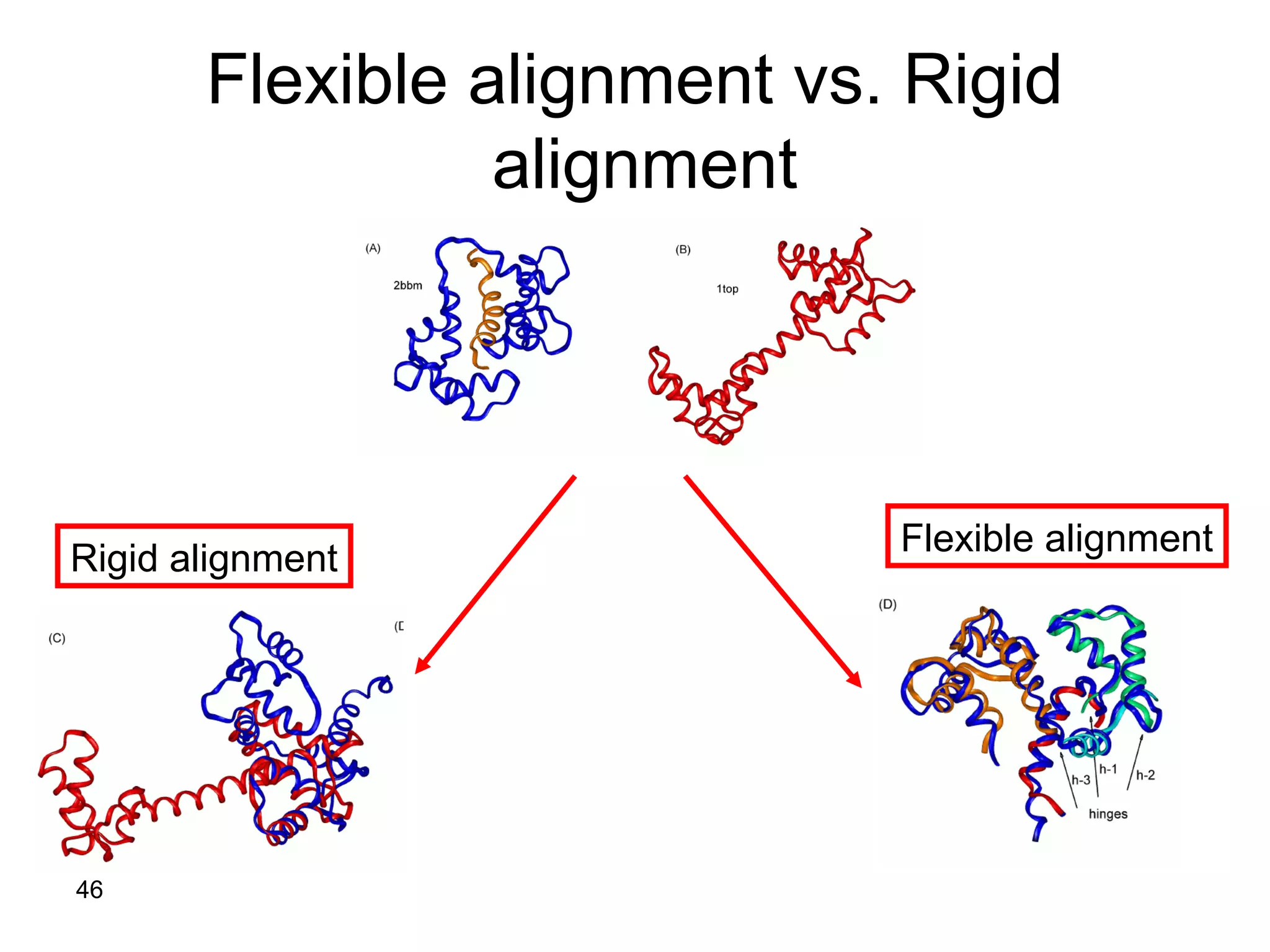
Protein structures can be aligned and compared using computational methods like structural alignment. Structural alignment finds the optimal rotation and translation that superimposes one protein structure onto another to maximize structural similarity. This is done by treating protein structures as sets of points defined by atom coordinates and finding the transformation that minimizes the root-mean-square deviation between corresponding atoms in the two structures. While useful, structural alignment has limitations like not accounting for differences in amino acid attributes and treating all atoms equally.