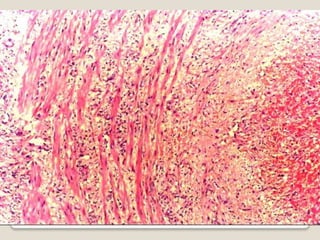
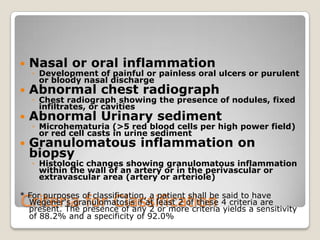
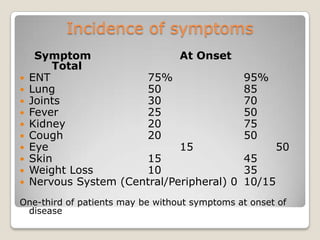
Vasculitis refers to inflammation of blood vessels. This document discusses the classification of various types of vasculitis based on vessel size, etiology, and other factors. Some of the main types covered include giant cell arteritis (temporal arteritis), Takayasu arteritis, polyarteritis nodosa, Buerger's disease, Kawasaki disease, microscopic polyangiitis, Churg-Strauss syndrome, and Wegener's granulomatosis. Key distinguishing clinical features, pathogenesis, diagnostic criteria and histopathological findings are provided for each condition.