Validation Master Plan (VMP) in Pharmaceutical.pptx
1.
Validation Master Plan(VMP) in
Pharmaceutical Industry
AN OVERVIEW OF THE VALIDATION MASTER
PLAN – A KEY DOCUMENT IN ENSURING
GMP COMPLIANCE.
Dr Seema A Gosavi
Associate Professor
Department of Pharmaceutical Chemistry
Sanjivani College of Pharmaceutical Education and Research, Kopargaon
E-mail: seemagosavibpharm@sanjivani.org.in
Mobile: 9860077084
2.
Learning Objectives:
• Understandthe concept and importance of the Validation Master
Plan (VMP).
• Identify the key components, roles, and responsibilities
associated with a VMP.
• Explain the validation lifecycle and associated documentation.
• Recognize the regulatory requirements and compliance aspects.
3.
Learning Outcomes:
Studentswill be able to define and describe the purpose of a VMP.
Students will identify and explain various types of validation (equipment,
process, cleaning, etc.).
Students will interpret the structure of a VMP and list key regulatory
references.
Students will evaluate the impact of validation failures and propose
mitigation strategies.
4.
Introduction
Validation isa fundamental requirement in pharmaceutical manufacturing.
Ensures consistent quality, safety, and efficacy of products.
VMP is the backbone of validation activities.
Definition:
A Validation Master Plan (VMP) is a comprehensive document that outlines the
overall strategy and approach for validation activities within a pharmaceutical
manufacturing facility. It serves as a roadmap for ensuring that all processes,
equipment, and systems meet predetermined quality standards and regulatory
requirements.
5.
What is aValidation Master Plan?
• A high-level document describing the company’s
approach to validation.
• Defines the scope, strategy, and responsibilities.
• Covers facilities, systems, equipment, and processes.
6.
Purpose of VMP
•Ensure product quality and regulatory compliance
• Provide structured validation activities
• Demonstrate GMP adherence
• To provide a framework for all validation activities.
• To define responsibilities, timelines, and scope of validation.
• To ensure compliance with regulatory guidelines (e.g., FDA,
EMA, WHO, PIC/S).
• To ensure product quality, safety, and efficacy through validated
processes.
• To coordinate validation efforts and avoid duplication.
7.
Scope of theVMP
Includes Facilities, Utilities, Equipment, Cleaning,
Computer Systems, And Analytical Methods That Affect
Product Quality.
• Defines which systems, equipment, processes, and utilities are covered.
• May include production, packaging, cleaning, utilities, computerized systems,
etc.
• Manufacturing processes
• Facilities and utilities
• Equipment and software systems
• Cleaning and analytical methods
8.
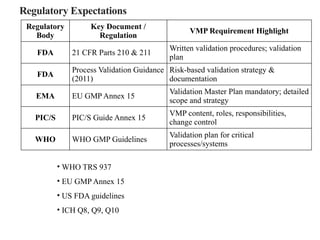
Regulatory Expectations
• WHOTRS 937
• EU GMP Annex 15
• US FDA guidelines
• ICH Q8, Q9, Q10
Regulatory
Body
Key Document /
Regulation
VMP Requirement Highlight
FDA 21 CFR Parts 210 & 211
Written validation procedures; validation
plan
FDA
Process Validation Guidance
(2011)
Risk-based validation strategy &
documentation
EMA EU GMP Annex 15
Validation Master Plan mandatory; detailed
scope and strategy
PIC/S PIC/S Guide Annex 15
VMP content, roles, responsibilities,
change control
WHO WHO GMP Guidelines
Validation plan for critical
processes/systems
9.
Key Components ofVMP
Component Description
1. Introduction
Overview of the facility, manufacturing processes, and purpose of
the VMP.
2. Scope
Defines which systems, processes, equipment, and utilities are
covered under the VMP.
3. Validation Policy
Company’s overall approach and commitment to validation, aligned
with regulatory requirements.
4. Organizational Structure
and Responsibilities
Roles and responsibilities of the validation team and involved
departments (QA, Production, etc.).
5. Validation Strategy
Description of validation types (process, equipment, cleaning,
computerized systems) and risk-based approach.
6. Validation Lifecycle and
Documentation
Stages of validation (DQ, IQ, OQ, PQ), protocol and report
requirements, documentation control.
7. Schedule and Timeline
Timeline and prioritization for validation activities, including project
milestones.
8. Change Control
Procedures for managing changes impacting validated systems and
when revalidation is necessary.
9. Training
Training requirements and plans for personnel involved in validation
activities.
10. Deviation and CAPA
Management
Handling deviations during validation and implementing
corrective/preventive actions.
11. References
List of regulatory guidelines, SOPs, and standards referenced in the
VMP.
10.
Structure of aVMP Document
Introduction and validation policy
Organizational responsibilities
Validation strategy and schedule
Documentation and reporting
11.
Validation Policy
Definition:
The ValidationPolicy is a formal statement that outlines the company’s commitment
and approach to validation. It sets the foundation for all validation activities and
ensures alignment with regulatory requirements and industry best practices.
Purpose of the Validation Policy:
• To establish a clear and consistent approach to validation across the organization.
• To ensure that all processes, equipment, systems, and methods affecting product
quality are validated.
• To comply with regulatory expectations and good manufacturing practices (GMP).
• To support product quality, safety, and efficacy through effective validation.
12.
Roles and Responsibilities
•QualityAssurance: Oversight and approvals
•Engineering: Equipment validation
•Manufacturing: Process knowledge and SOP adherence
Role Responsibilities
Validation
Manager
- Develops and maintains the VMP - Oversees all validation activities -
Approves protocols/reports
Quality
Assurance (QA)
- Reviews/approves VMP, protocols, and reports - Ensures compliance
with GxP and regulatory guidelines - Manages deviations and CAPAs
Validation
Engineer
- Drafts and executes validation protocols (IQ, OQ, PQ) - Records
results - Investigates deviations
Process Owner /
Dept. Head
- Provides technical input for validation - Ensures validated systems
are used properly - Supports re-validation efforts
Engineering /
Maintenance
- Installs/calibrates equipment - Provides technical documentation -
Supports validation and change control
Regulatory
Affairs
- Ensures validation aligns with regulations - Supports inspections and
submissions with validation data
IT / CSV Team
- Validates computerized systems - Ensures 21 CFR Part 11 and data
integrity compliance - Maintains system validation
13.
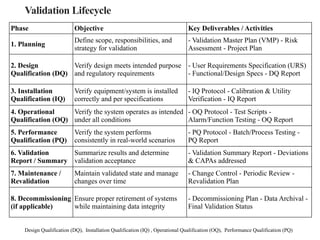
Validation Lifecycle
Design Qualification(DQ), Installation Qualification (IQ) , Operational Qualification (OQ), Performance Qualification (PQ)
Phase Objective Key Deliverables / Activities
1. Planning
Define scope, responsibilities, and
strategy for validation
- Validation Master Plan (VMP) - Risk
Assessment - Project Plan
2. Design
Qualification (DQ)
Verify design meets intended purpose
and regulatory requirements
- User Requirements Specification (URS)
- Functional/Design Specs - DQ Report
3. Installation
Qualification (IQ)
Verify equipment/system is installed
correctly and per specifications
- IQ Protocol - Calibration & Utility
Verification - IQ Report
4. Operational
Qualification (OQ)
Verify the system operates as intended
under all conditions
- OQ Protocol - Test Scripts -
Alarm/Function Testing - OQ Report
5. Performance
Qualification (PQ)
Verify the system performs
consistently in real-world scenarios
- PQ Protocol - Batch/Process Testing -
PQ Report
6. Validation
Report / Summary
Summarize results and determine
validation acceptance
- Validation Summary Report - Deviations
& CAPAs addressed
7. Maintenance /
Revalidation
Maintain validated state and manage
changes over time
- Change Control - Periodic Review -
Revalidation Plan
8. Decommissioning
(if applicable)
Ensure proper retirement of systems
while maintaining data integrity
- Decommissioning Plan - Data Archival -
Final Validation Status
14.
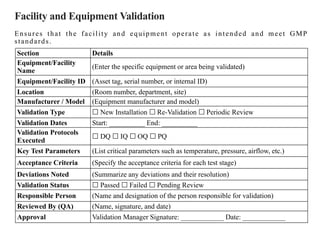
Facility and EquipmentValidation
Ensures that the facility and equipment operate as intended and meet GMP
standards.
Section Details
Equipment/Facility
Name
(Enter the specific equipment or area being validated)
Equipment/Facility ID (Asset tag, serial number, or internal ID)
Location (Room number, department, site)
Manufacturer / Model (Equipment manufacturer and model)
Validation Type ☐ New Installation Re-Validation Periodic Review
☐ ☐
Validation Dates Start: __________ End: __________
Validation Protocols
Executed
☐ DQ IQ OQ PQ
☐ ☐ ☐
Key Test Parameters (List critical parameters such as temperature, pressure, airflow, etc.)
Acceptance Criteria (Specify the acceptance criteria for each test stage)
Deviations Noted (Summarize any deviations and their resolution)
Validation Status ☐ Passed Failed Pending Review
☐ ☐
Responsible Person (Name and designation of the person responsible for validation)
Reviewed By (QA) (Name, signature, and date)
Approval Validation Manager Signature: ____________ Date: ____________
15.
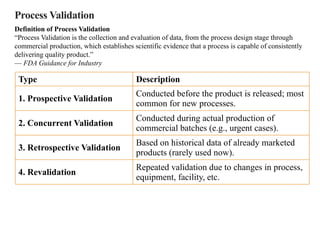
Process Validation
Definition ofProcess Validation
“Process Validation is the collection and evaluation of data, from the process design stage through
commercial production, which establishes scientific evidence that a process is capable of consistently
delivering quality product.”
— FDA Guidance for Industry
Type Description
1. Prospective Validation
Conducted before the product is released; most
common for new processes.
2. Concurrent Validation
Conducted during actual production of
commercial batches (e.g., urgent cases).
3. Retrospective Validation
Based on historical data of already marketed
products (rarely used now).
4. Revalidation
Repeated validation due to changes in process,
equipment, facility, etc.
16.
In a ValidationMaster Plan
(VMP), process validation is a
core section that outlines the
strategy, scope, and
responsibilities for validating
pharmaceutical manufacturing
processes. It provides a high-
level roadmap to ensure
consistent product quality and
compliance with regulatory
requirements (e.g., FDA,
EMA, WHO, ICH Q8-Q10).
Purpose of Including Process
Validation in VMP
Ensure all critical processes
are validated before commercial
production.
Establish a standardized
approach for process
qualification.
Meet regulatory expectations
for lifecycle validation.
Provide traceability and
control across all stages of
validation.
Process Validation
17.
Key Elements ofProcess Validation in a VMP
Section Content Description
Objective Define the purpose of process validation and its role in assuring product quality.
Scope
Covers all manufacturing processes, including critical steps and control
strategies.
Regulatory
References
ICH Q8-Q10, FDA Guidance (2011), EU Annex 15, WHO TRS, etc.
Validation Approach
Lifecycle-based approach (Process Design, Qualification, Continued
Verification).
Types of Validation Prospective, Concurrent, Retrospective (if justified), and Revalidation.
Process Qualification
Strategy
Outline how many batches will be validated, sampling plan, and statistical
justification.
Roles and
Responsibilities
QA, Production, Validation team, Engineering—clearly defined in a RACI
matrix.
Documentation Process validation protocols, reports, batch records, deviations, CAPAs.
Change Control Criteria for revalidation in case of process changes.
Risk Assessment
Application of Quality Risk Management (QRM) to determine validation scope
and depth.
Continued Process
Verification
Plan for ongoing monitoring (e.g., trending, statistical process control).
18.
Establishes documented evidencethat cleaning procedures remove
residues to an acceptable level.
Cleaning Validation in the Validation Master Plan (VMP)
Cleaning Validation is a crucial section of the Validation Master Plan (VMP) in
pharmaceutical manufacturing. It ensures that cleaning procedures effectively remove
product residues, cleaning agents, and contaminants from equipment to prevent cross-
contamination and ensure product quality.
Purpose of Cleaning Validation in the VMP
•Demonstrate that cleaning processes consistently meet predefined acceptance criteria.
•Ensure compliance with regulatory requirements (FDA, EU GMP, WHO, PIC/S).
•Prevent cross-contamination and ensure patient safety.
19.
Key Elements ofCleaning Validation in the VMP
Section Content
Objective Define the purpose of cleaning validation within the overall validation strategy.
Scope Identify which equipment, products, and cleaning procedures are covered.
Regulatory
References
FDA 21 CFR Part 211, EU Annex 15, WHO TRS 937, PIC/S PI 006, ICH Q9.
Cleaning Strategies
- Product-specific vs. campaign-based cleaning - Manual vs. automated (CIP/SIP)
methods
Worst-Case
Selection
Justification of worst-case product/equipment for validation studies.
Acceptance Criteria
- Residue limits (MACO – Maximum Allowable Carry Over) - Swab and rinse
limits - Microbial & endotoxin limits
Sampling Methods - Swab sampling - Rinse sampling - Visual inspection
Analytical Methods - Sensitivity and specificity of residue detection - LOD/LOQ validated methods
Number of Runs Typically three consecutive successful cleaning runs to validate the process.
Revalidation
Requirements
When to revalidate (e.g., changes in product, equipment, cleaning procedure).
Roles and
Responsibilities
QA, Production, Validation, QC – defined via RACI or responsibility matrix.
Documentation
Required
- Cleaning validation protocols and reports - SOPs - Equipment logs
20.
Computer System Validation
EnsuresSoftware And Hardware Systems Perform Accurately And Reliably
In Compliance With 21 CFR Part 11.
Computer System Validation (CSV) is a mandatory part of the Validation Master Plan
(VMP) for regulated pharmaceutical environments. It ensures that computerized systems
used in GxP processes are reliable, accurate, secure, and compliant with regulatory
requirements such as 21 CFR Part 11, EU Annex 11, and GAMP 5.
Purpose of Including CSV in the VMP
• Ensure data integrity, security, and traceability.
• Confirm computerized systems function as intended and meet user requirements.
• Establish a structured lifecycle validation approach.
• Maintain compliance with regulatory expectations (FDA, EMA, MHRA, etc.).
21.
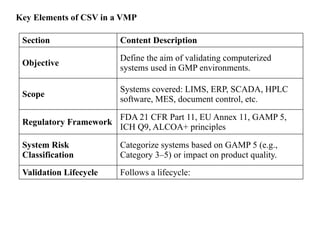
Key Elements ofCSV in a VMP
Section Content Description
Objective
Define the aim of validating computerized
systems used in GMP environments.
Scope
Systems covered: LIMS, ERP, SCADA, HPLC
software, MES, document control, etc.
Regulatory Framework
FDA 21 CFR Part 11, EU Annex 11, GAMP 5,
ICH Q9, ALCOA+ principles
System Risk
Classification
Categorize systems based on GAMP 5 (e.g.,
Category 3–5) or impact on product quality.
Validation Lifecycle Follows a lifecycle:
22.
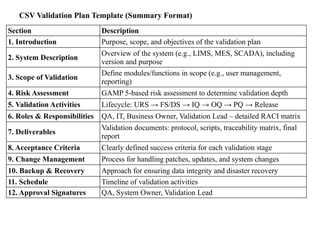
CSV Validation PlanTemplate (Summary Format)
Section Description
1. Introduction Purpose, scope, and objectives of the validation plan
2. System Description
Overview of the system (e.g., LIMS, MES, SCADA), including
version and purpose
3. Scope of Validation
Define modules/functions in scope (e.g., user management,
reporting)
4. Risk Assessment GAMP 5-based risk assessment to determine validation depth
5. Validation Activities Lifecycle: URS → FS/DS → IQ → OQ → PQ → Release
6. Roles & Responsibilities QA, IT, Business Owner, Validation Lead – detailed RACI matrix
7. Deliverables
Validation documents: protocol, scripts, traceability matrix, final
report
8. Acceptance Criteria Clearly defined success criteria for each validation stage
9. Change Management Process for handling patches, updates, and system changes
10. Backup & Recovery Approach for ensuring data integrity and disaster recovery
11. Schedule Timeline of validation activities
12. Approval Signatures QA, System Owner, Validation Lead
23.
Traceability Matrix Template(CSV)
URS ID
Functional
Requirement
Test Case
ID
Test
Description
Test
Result
Remarks
URS-01
User login must
be secure
TC-01
Verify
password
complexity
rules
Pass/Fail
URS-02
Audit trail
functionality
TC-02
Check system
logs for
changes
Pass/Fail
24.
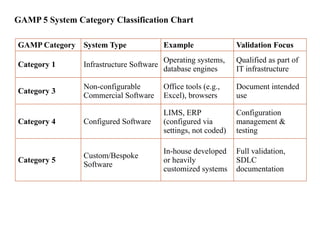
GAMP 5 SystemCategory Classification Chart
GAMP Category System Type Example Validation Focus
Category 1 Infrastructure Software
Operating systems,
database engines
Qualified as part of
IT infrastructure
Category 3
Non-configurable
Commercial Software
Office tools (e.g.,
Excel), browsers
Document intended
use
Category 4 Configured Software
LIMS, ERP
(configured via
settings, not coded)
Configuration
management &
testing
Category 5
Custom/Bespoke
Software
In-house developed
or heavily
customized systems
Full validation,
SDLC
documentation
25.
Analytical Method Validation
Confirmsthat analytical methods are suitable for their intended
use in terms of accuracy, precision, specificity, and robustness.
Analytical Method Validation (AMV) is a key component of the Validation
Master Plan (VMP) in the pharmaceutical industry. It ensures that the
analytical methods used for testing raw materials, intermediates, and finished
products are scientifically sound, reproducible, and suitable for their
intended purpose.
Purpose of AMV in VMP
•Confirm methods provide accurate, reliable, and consistent results.
•Ensure compliance with regulatory expectations:
• ICH Q2(R2) (newest)
• USP <1225>, <1226>
• EMA, FDA, WHO TRS
26.
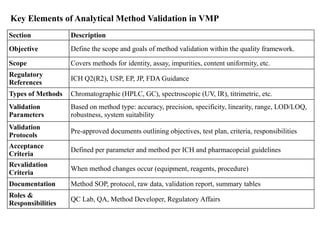
Key Elements ofAnalytical Method Validation in VMP
Section Description
Objective Define the scope and goals of method validation within the quality framework.
Scope Covers methods for identity, assay, impurities, content uniformity, etc.
Regulatory
References
ICH Q2(R2), USP, EP, JP, FDA Guidance
Types of Methods Chromatographic (HPLC, GC), spectroscopic (UV, IR), titrimetric, etc.
Validation
Parameters
Based on method type: accuracy, precision, specificity, linearity, range, LOD/LOQ,
robustness, system suitability
Validation
Protocols
Pre-approved documents outlining objectives, test plan, criteria, responsibilities
Acceptance
Criteria
Defined per parameter and method per ICH and pharmacopeial guidelines
Revalidation
Criteria
When method changes occur (equipment, reagents, procedure)
Documentation Method SOP, protocol, raw data, validation report, summary tables
Roles &
Responsibilities
QC Lab, QA, Method Developer, Regulatory Affairs
27.
Typical Validation Parameters(ICH Q2)
Parameter Applicable To
Specificity All methods
Linearity Quantitative methods
Accuracy Quantitative methods
Precision Repeatability and intermediate
Detection Limit Impurities and limit tests
Quantitation Limit Assay and impurity methods
Robustness All methods
System Suitability All methods with instrumentation
28.
Revalidation
Performed when changesoccur or after a defined period to ensure
continued compliance and performance.
Revalidation ensures that validated systems, equipment, and processes remain in
a state of control over time. It is a core element of the Validation Master Plan
(VMP) to maintain compliance and product quality in pharmaceutical
manufacturing.
Purpose of Revalidation
• To confirm that previously validated systems still meet predefined criteria.
• Required when changes, deviations, or time-based intervals occur.
• Ensures continued assurance of process performance and product quality.
29.
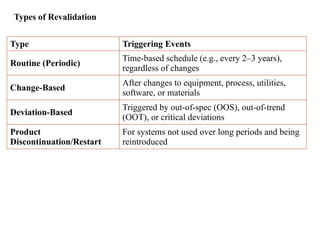
Types of Revalidation
TypeTriggering Events
Routine (Periodic)
Time-based schedule (e.g., every 2–3 years),
regardless of changes
Change-Based
After changes to equipment, process, utilities,
software, or materials
Deviation-Based
Triggered by out-of-spec (OOS), out-of-trend
(OOT), or critical deviations
Product
Discontinuation/Restart
For systems not used over long periods and being
reintroduced
30.
Scope of Revalidationin VMP
Area Revalidation Requirement
Facilities & HVAC Structural modifications, new installations
Equipment Relocation, major maintenance, software upgrades
Utilities (Water, Air,
Steam)
Changes to generation/distribution systems
Process
Major changes to critical parameters, raw materials,
batch size
Cleaning Procedures
Detergent change, new contact surface, failure in
cleaning verification
Computer Systems Software version updates, configuration changes
Analytical Methods Modified procedures, new instruments
31.
Key VMP DeliverablesRelated to Revalidation
• Revalidation Schedule (calendar or tracker)
• Change Control Records
• Risk Assessment Reports
• Revalidation Protocols & Reports
• Deviation & Investigation Reports
• Final Approval and QA Summary
32.
Change Control
Change Controlis a formal system used to manage, document, and evaluate
any changes that could impact the validated state of systems, processes,
equipment, or facilities. It ensures product quality, safety, and regulatory
compliance are maintained.
Objective of Change Control in VMP
To establish a structured process to:
• Assess proposed changes.
• Evaluate risks and impact.
• Implement changes with necessary revalidation.
• Maintain traceability and compliance.
33.
Key Elements ofChange Control in the VMP
Component Description
Scope
Includes changes to equipment, processes, software, documents,
and utilities
Initiation
Change is proposed and documented via a change control form
(CCF)
Impact
Assessment
Cross-functional risk evaluation (impact on quality, safety,
validation status)
Change
Classification
Minor, Major, or Critical (based on risk to validated state or
compliance)
Review &
Approval
QA, Validation, and Functional Owners must review and approve
Implementati
on Plan
Includes actions, testing, training, and timelines
Revalidation
Decision
Based on risk and impact; documented in the change control record
Closure Verified post-implementation; QA confirms all actions completed
RiskAssessment in VMP
RiskAssessment in the VMP provides a structured approach to
identify, evaluate, and mitigate potential risks that may affect
product quality, patient safety, and regulatory compliance during
validation activities.
Purpose of Risk Assessment in VMP
•To apply science- and risk-based principles (aligned with ICH Q9).
•To determine the scope and extent of validation activities.
•To prioritize efforts based on risk level (High, Medium, Low).
•To support change control, deviation, and revalidation decisions.
36.
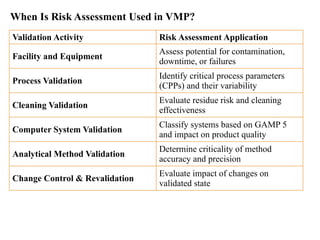
When Is RiskAssessment Used in VMP?
Validation Activity Risk Assessment Application
Facility and Equipment
Assess potential for contamination,
downtime, or failures
Process Validation
Identify critical process parameters
(CPPs) and their variability
Cleaning Validation
Evaluate residue risk and cleaning
effectiveness
Computer System Validation
Classify systems based on GAMP 5
and impact on product quality
Analytical Method Validation
Determine criticality of method
accuracy and precision
Change Control & Revalidation
Evaluate impact of changes on
validated state
37.
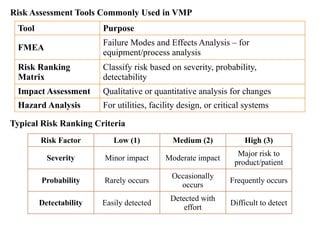
Risk Assessment ToolsCommonly Used in VMP
Tool Purpose
FMEA
Failure Modes and Effects Analysis – for
equipment/process analysis
Risk Ranking
Matrix
Classify risk based on severity, probability,
detectability
Impact Assessment Qualitative or quantitative analysis for changes
Hazard Analysis For utilities, facility design, or critical systems
Typical Risk Ranking Criteria
Risk Factor Low (1) Medium (2) High (3)
Severity Minor impact Moderate impact
Major risk to
product/patient
Probability Rarely occurs
Occasionally
occurs
Frequently occurs
Detectability Easily detected
Detected with
effort
Difficult to detect
38.
Validation Planning
Validation Planningin a VMP outlines how validation activities will be
structured, executed, and documented to ensure compliance with regulatory
expectations (e.g., FDA, EMA, WHO).
Key Components of Validation Planning in VMP
Component Description
Scope of Validation
Defines systems, processes, and equipment included in the validation
effort
Validation Strategy Approach (e.g., prospective, concurrent, retrospective) based on risk
Validation Schedule Timeline or Gantt chart outlining validation activities
Resources Required Teams, departments, and third parties involved
Documentation Plan
Required protocols and reports (e.g., URS, DQ, IQ, OQ, PQ, summary
reports)
Roles &
Responsibilities
Assignment of responsibility for approvals, execution, review
Acceptance Criteria Clear, measurable criteria to determine if validation is successful
Deviation Management Procedures to handle unexpected results or deviations during validation
39.
Example: Validation Timeline
MonthActivity
Jan User Requirements Specification (URS)
Feb–Mar
Design Qualification (DQ), Installation Qualification
(IQ)
Apr–May Operational Qualification (OQ)
Jun Performance Qualification (PQ)
Jul Summary Report & Final Review
40.
Document Control
Document Controlensures that all validation-related documents are properly created,
reviewed, approved, issued, distributed, revised, and archived in a controlled manner.
Purpose in VMP
To maintain the integrity, traceability, and regulatory compliance of all documents
generated during validation activities.
Key Elements of Document Control
Aspect Description
Document Numbering Unique ID assigned to each validation document for traceability
Version Control Tracks changes and maintains current approved versions
Approval Process Review and sign-off by QA, Validation, and other stakeholders
Distribution Ensures only the current version is available for use
Archiving & Retention Secure storage and retrieval for a defined retention period
Access Control Limits editing/approval rights to authorized personnel
Change History (Audit
Trail)
Records all document modifications with date and responsible person
41.
Examples of ControlledDocuments
•Validation Master Plan (VMP)
•Validation Protocols (IQ, OQ, PQ)
•Risk Assessments
•Traceability Matrix
•Deviation Reports
•Summary Reports
•SOPs related to validation
Best Practices
•Use a Document Management System
(DMS) or Electronic Document
Management System (EDMS).
•Implement periodic reviews and revisions
based on product lifecycle or regulatory
updates.
•Include a Document Control SOP in the
VMP.
42.
Training Requirements ina Validation Master Plan
Purpose of Training Requirements
To ensure that all personnel involved in validation activities:
•Understand their roles and responsibilities.
•Possess adequate knowledge of GMP (Good Manufacturing Practice).
•Are competent in using relevant equipment, procedures, and documentation practices.
Who Needs to be Trained?
• Validation team members
• QA/QC personnel
• Production and maintenance staff
• Engineering teams
• Data integrity and documentation personnel
Types of Required Training
• GMP & Regulatory Compliance Training
• Validation Principles & Protocols (IQ, OQ, PQ)
• Equipment Qualification Procedures
• Cleaning Validation
• Analytical Method Validation
• Software/Computer System Validation
• Deviation and Change Control Handling
Training Methodologies
• Classroom sessions
• On-the-job training
• SOP-based instructions
• External workshops/seminars
• E-learning modules (for periodic refreshers)
43.
Training Documentation
•Training recordsmust be maintained as per GDP.
•Each record should include:
• Date of training
• Trainer and trainee names
• Content or SOPs covered
• Evaluation results (if any)
• Signatures for acknowledgment
Evaluation of Training Effectiveness
•Post-training assessments (quizzes or practical demos)
•Supervisor observation and feedback
•Periodic re-evaluation (e.g., annually)
Retraining Triggers
•SOP or regulatory changes
•Equipment upgrades
•After audits or non-compliance findings
•Observed errors or incidents
Responsibilities
•QA Department: Ensures training needs are
identified and met.
•HR/Training Coordinator: Schedules and
tracks training programs.
•Supervisors: Ensure team participation and
effectiveness.
•Employees: Attend, understand, and
implement training.
44.
Training Requirements –Summary Table for VMP
Element Details
Objective Ensure personnel are qualified to perform validation activities.
Personnel to be
Trained
Validation team, QA/QC, production, engineering, maintenance, IT
staff.
Training Topics
GMP, IQ/OQ/PQ, equipment qualification, cleaning validation, data
integrity.
Training Methods Classroom, on-the-job, SOP-based, e-learning, external workshops.
Training Frequency
Initial (induction), periodic (e.g., annually), as needed (SOP
changes).
Evaluation Methods Written tests, practical demonstrations, supervisor evaluations.
Training Records
Maintained as per GDP; includes date, content, signatures,
evaluations.
Retraining Triggers SOP changes, audit findings, incidents, equipment changes.
Responsible
Departments
QA, HR/Training Coordinator, Supervisors.
45.
QualityAssurance Role –Summary Table
QA Responsibility Details
Review & Approve VMP, protocols (IQ/OQ/PQ), validation reports
Compliance Oversight Ensures validation aligns with GMP and regulatory standards
Documentation
Control
Manages SOPs, validation records, and training documents
Deviation/CAPA
Management
Handles validation deviations and verifies effectiveness of
CAPA
Risk Assessment Participates in validation risk analysis and mitigation planning
Training Verification Ensures all validation personnel are adequately trained
Audit Participation Prepares for internal/external audits, addresses findings
Continuous
Improvement
Suggests improvements in validation based on audit trends and
feedback
46.
Monitoring and Review
Objective:
Toestablish a structured approach for continuous
oversight and periodic evaluation of validation
activities to ensure sustained compliance and
effectiveness.
Key Components
1. Routine Monitoring
•Regular tracking of validation progress against the VMP schedule.
•Monitoring of validation metrics like completion rates, deviations, CAPA
closure, etc.
•Evaluation of system performance post-validation to detect drift or non-
conformance.
2. Review Frequency
•Typically conducted quarterly or annually depending on the scope and
risk.
•Triggered by events such as:
• Change controls
• Deviations
• Audit observations
• Product quality complaints
3. Review Activities
•Review of:
• Completed validation protocols and reports
• Open and overdue validation items
• Deviations and CAPAs
• Training records
•Comparison with regulatory expectations and industry best practices.
4. Outcomes of Review
•Identification of areas needing
revalidation.
•Recommendations for changes in
procedures, training, or documentation.
•Update or revision of the VMP if
necessary.
•Enhanced planning for future validations.
5. Roles and Responsibilities
•QA Department: Leads the review,
records findings, initiates necessary
actions.
•Validation Team: Provides
documentation and evidence.
•Management: Approves any VMP
updates or resource reallocations.
47.
Monitoring and Review– Summary Table
Aspect Details
Purpose Ensure validation remains effective and compliant
Frequency
Quarterly, annually, or based on events (e.g.,
deviations, audits)
What is Monitored?
Validation timelines, deviations, CAPAs,
performance, training, compliance
Key Activities
Document review, gap analysis, trend analysis, KPI
tracking
Responsible Parties QA (lead), validation team, management
Outcomes
Revalidation, updates to VMP, improved training or
process controls
48.
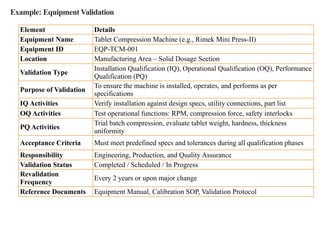
Example: Equipment Validation
ElementDetails
Equipment Name Tablet Compression Machine (e.g., Rimek Mini Press-II)
Equipment ID EQP-TCM-001
Location Manufacturing Area – Solid Dosage Section
Validation Type
Installation Qualification (IQ), Operational Qualification (OQ), Performance
Qualification (PQ)
Purpose of Validation
To ensure the machine is installed, operates, and performs as per
specifications
IQ Activities Verify installation against design specs, utility connections, part list
OQ Activities Test operational functions: RPM, compression force, safety interlocks
PQ Activities
Trial batch compression, evaluate tablet weight, hardness, thickness
uniformity
Acceptance Criteria Must meet predefined specs and tolerances during all qualification phases
Responsibility Engineering, Production, and Quality Assurance
Validation Status Completed / Scheduled / In Progress
Revalidation
Frequency
Every 2 years or upon major change
Reference Documents Equipment Manual, Calibration SOP, Validation Protocol
49.
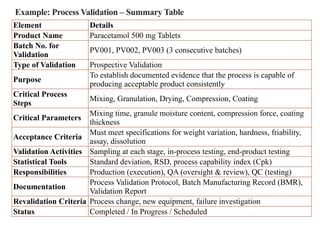
Example: Process Validation– Summary Table
Element Details
Product Name Paracetamol 500 mg Tablets
Batch No. for
Validation
PV001, PV002, PV003 (3 consecutive batches)
Type of Validation Prospective Validation
Purpose
To establish documented evidence that the process is capable of
producing acceptable product consistently
Critical Process
Steps
Mixing, Granulation, Drying, Compression, Coating
Critical Parameters
Mixing time, granule moisture content, compression force, coating
thickness
Acceptance Criteria
Must meet specifications for weight variation, hardness, friability,
assay, dissolution
Validation Activities Sampling at each stage, in-process testing, end-product testing
Statistical Tools Standard deviation, RSD, process capability index (Cpk)
Responsibilities Production (execution), QA (oversight & review), QC (testing)
Documentation
Process Validation Protocol, Batch Manufacturing Record (BMR),
Validation Report
Revalidation Criteria Process change, new equipment, failure investigation
Status Completed / In Progress / Scheduled
50.
Example: Cleaning Validation– Summary Table
Element Details
Equipment Name Fluid Bed Dryer (FBD)
Product Cleaned From Metformin Hydrochloride Tablets
Next Product Paracetamol Tablets
Cleaning Method
Manual cleaning using water and approved detergent, followed
by drying
Type of Validation Prospective / Concurrent / Periodic
Sampling Methods Swab sampling & Rinse sampling
Acceptance Criteria
<10 ppm product residue, <100 CFU/ml microbial count, no
visible residue
Worst Case
Determination
Based on product solubility, potency, batch size, difficulty to
clean
Analytical Method Used HPLC for residue analysis, Bioburden for microbial limits
No. of Validation Runs 3 consecutive successful cleanings
Locations Sampled Hard-to-clean surfaces: corners, joints, outlet chute, filter housing
Documentation
Cleaning Validation Protocol, Sample Plan, Test Results, Final
Report
Revalidation Criteria
Change in cleaning procedure, new product introduction,
equipment modification
Status Completed / Scheduled / In Progress
51.
Deviation Management
Objective ofDeviation Management in VMP:
• To establish a standardized and systematic approach for identifying, documenting, evaluating, and
resolving deviations encountered during validation activities.
• To ensure that all deviations are managed in compliance with regulatory requirements and do not
compromise product quality, safety, or efficacy.
• To ensure that appropriate corrective and preventive actions (CAPA) are implemented and monitored
effectively.
Purpose of Deviation Management in VMP:
• To maintain control over validation processes by effectively addressing deviations from approved
procedures or specifications.
• To ensure transparency and traceability in handling non-conformities during validation.
• To enable root cause analysis and continuous improvement through proper investigation and
trending of deviations.
• To minimize the risk of product defects, regulatory non-compliance, and process variability due to
unaddressed deviations.
52.
Deviation Management inValidation Master Plan (VMP)
Aspect Description
Definition
A deviation is any departure from an approved process, instruction, or standard operating
procedure (SOP) during validation.
Purpose
To ensure all deviations are properly identified, documented, investigated, and resolved
to maintain product quality and compliance.
Types of Deviations
- Planned Deviation: Authorized change from a procedure with prior approval.-
Unplanned Deviation: Unexpected departure during execution.
Deviation
Identification
Any personnel involved in validation activities can report a deviation immediately upon
noticing.
Documentation
All deviations must be recorded in a deviation report with detailed description, date/time,
location, and responsible personnel.
Investigation Process
Root cause analysis (RCA) conducted to determine underlying cause(s). Tools: 5 Whys,
Fishbone Diagram, etc.
Impact Assessment
Evaluate deviation impact on:- Product quality- Validation outcome- Regulatory
compliance
Corrective &
Preventive Actions
(CAPA)
- Corrective Action: Fix the issue.- Preventive Action: Prevent recurrence.
Approval & Closure
QA/Validation Head reviews and approves closure of the deviation after CAPA
implementation.
Revalidation Trigger
Significant deviations may trigger full or partial revalidation depending on the severity
and impact.
Trending &
Monitoring
Periodic review of deviation data to identify trends or repeated issues, improving future
processes.
53.
Validation Report
Objective ofValidation Report in VMP:
•To formally document and summarize the outcome of a validation activity.
•To verify that all validation steps were completed according to the protocol and met pre-
defined acceptance criteria.
•To ensure that the validated process, equipment, or system performs consistently and
reproducibly within established specifications.
Purpose of Validation Report in VMP:
•To provide documented evidence that the system or process meets its intended use and
regulatory requirements.
•To serve as a comprehensive record for internal audits and external inspections.
•To support decisions regarding the approval, continuation, or revalidation of equipment,
processes, or systems.
•To include deviations, investigations, CAPAs, and conclusions related to the validation
exercise.
54.
Validation Report inVMP – Summary Table
Section Description
Objective Summarizes the purpose and scope of the validation study
System/Process Validated
Name and description of the system, equipment, process, or
method validated
Validation Type
Type of validation (e.g., Installation, Operational, Performance,
Cleaning)
Protocol Reference Identification number and title of the validation protocol used
Execution Summary
Summary of test execution, including dates and involved
personnel
Results Summary Overview of test results: passed/failed with justification
Deviations
Description of any deviations encountered and corrective
actions taken
Acceptance Criteria Whether acceptance criteria were met
Conclusions
Final conclusion about the state of validation (e.g.,
successful/failed)
Recommendations
Any recommendations for re-validation, preventive actions, or
improvements
Approval Signatures
QA, validation, and relevant department heads approval with
date
55.
Auditing and Inspections
Ina Validation Master Plan (VMP), the "Auditing and Inspections" section outlines how
validation processes are monitored and evaluated for compliance. Here's a structured
summary table:
Element Description
Objective
To verify that validation activities comply with internal procedures and
regulations
Types of Audits Internal audits, external audits, vendor audits, regulatory inspections
Audit Frequency
Defined periodicity (e.g., annual, biannual) depending on
system/process criticality
Responsible
Department
Quality Assurance (QA) or Compliance department
Audit Scope
Includes validation protocols, reports, data integrity, equipment logs,
etc.
Inspection Readiness Ensures all documentation is complete, traceable, and readily available
Follow-Up and CAPA Corrective and Preventive Actions to address audit findings
Documentation Audit reports, checklists, compliance certificates
Regulatory Bodies
Involved
Examples: FDA, EMA, WHO, local health authorities
Review and Approval Validation and QA management must review and approve audit findings
Auditing and Inspections in VMP – Summary Table
56.
Common VMPMistakes
Mistake DescriptionImpact
1. Vague Objectives
Lack of clear scope or purpose of the
validation activities
Leads to confusion and
inconsistent implementation
2. Incomplete System
Inventory
Missing list of all
systems/equipment/processes to be
validated
Critical systems may be excluded
from validation
3. Poor Risk
Assessment
No or inadequate risk-based approach to
validation priorities
Resources are misallocated; non-
critical systems overemphasized
4. Lack of Traceability
Missing links between user requirements,
test cases, and results
Compromises compliance and
audit readiness
5. Undefined Roles &
Responsibilities
No clarity on who does what in validation
Causes delays, accountability
issues, and poor execution
6. Outdated
References
Use of obsolete standards (e.g., old GAMP
or ICH guidelines)
Regulatory non-compliance
7. Weak Change
Control Integration
Change control not tied into the validation
lifecycle
Unauthorized changes can go
unvalidated
8. Inconsistent
Documentation
Practices
Inadequate version control, unsigned
documents, or missing approvals
Leads to audit findings and poor
data integrity
9. Missing
Revalidation Criteria
No guidance on when to revalidate after
changes or time periods
May result in operating non-
compliant systems
10. Poor Review &
Approval Process
VMP not formally reviewed or approved by
QA and management
Lack of ownership and oversight
57.
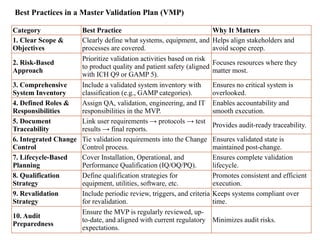
Best Practices ina Master Validation Plan (VMP)
Category Best Practice Why It Matters
1. Clear Scope &
Objectives
Clearly define what systems, equipment, and
processes are covered.
Helps align stakeholders and
avoid scope creep.
2. Risk-Based
Approach
Prioritize validation activities based on risk
to product quality and patient safety (aligned
with ICH Q9 or GAMP 5).
Focuses resources where they
matter most.
3. Comprehensive
System Inventory
Include a validated system inventory with
classification (e.g., GAMP categories).
Ensures no critical system is
overlooked.
4. Defined Roles &
Responsibilities
Assign QA, validation, engineering, and IT
responsibilities in the MVP.
Enables accountability and
smooth execution.
5. Document
Traceability
Link user requirements → protocols → test
results → final reports.
Provides audit-ready traceability.
6. Integrated Change
Control
Tie validation requirements into the Change
Control process.
Ensures validated state is
maintained post-change.
7. Lifecycle-Based
Planning
Cover Installation, Operational, and
Performance Qualification (IQ/OQ/PQ).
Ensures complete validation
lifecycle.
8. Qualification
Strategy
Define qualification strategies for
equipment, utilities, software, etc.
Promotes consistent and efficient
execution.
9. Revalidation
Strategy
Include periodic review, triggers, and criteria
for revalidation.
Keeps systems compliant over
time.
10. Audit
Preparedness
Ensure the MVP is regularly reviewed, up-
to-date, and aligned with current regulatory
expectations.
Minimizes audit risks.
58.
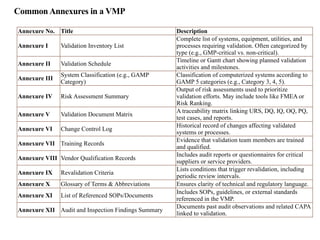
CommonAnnexures in aVMP
Annexure No. Title Description
Annexure I Validation Inventory List
Complete list of systems, equipment, utilities, and
processes requiring validation. Often categorized by
type (e.g., GMP-critical vs. non-critical).
Annexure II Validation Schedule
Timeline or Gantt chart showing planned validation
activities and milestones.
Annexure III
System Classification (e.g., GAMP
Category)
Classification of computerized systems according to
GAMP 5 categories (e.g., Category 3, 4, 5).
Annexure IV Risk Assessment Summary
Output of risk assessments used to prioritize
validation efforts. May include tools like FMEA or
Risk Ranking.
Annexure V Validation Document Matrix
A traceability matrix linking URS, DQ, IQ, OQ, PQ,
test cases, and reports.
Annexure VI Change Control Log
Historical record of changes affecting validated
systems or processes.
Annexure VII Training Records
Evidence that validation team members are trained
and qualified.
Annexure VIII Vendor Qualification Records
Includes audit reports or questionnaires for critical
suppliers or service providers.
Annexure IX Revalidation Criteria
Lists conditions that trigger revalidation, including
periodic review intervals.
Annexure X Glossary of Terms & Abbreviations Ensures clarity of technical and regulatory language.
Annexure XI List of Referenced SOPs/Documents
Includes SOPs, guidelines, or external standards
referenced in the VMP.
Annexure XII Audit and Inspection Findings Summary
Documents past audit observations and related CAPA
linked to validation.
59.
Regulatory Updates
The RegulatoryUpdates section of a VMP ensures that the validation strategy remains
aligned with current regulatory expectations, guidance, and industry best practices.
This is critical for maintaining compliance during inspections and audits.
Purpose of Including Regulatory Updates in VMP
•Demonstrate awareness and integration of current regulations and guidance.
•Ensure that validation activities reflect up-to-date regulatory requirements.
•Support risk-based approaches by linking them to the latest industry standards.
Best Practices
•Review and update this section annually or upon major regulatory changes.
•Link updates directly to validation SOPs or change controls.
•Maintain a Regulatory Change Log as an annexure.
60.
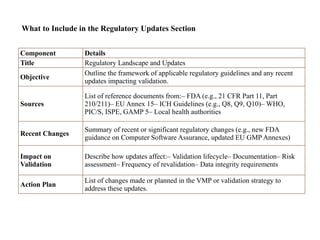
Component Details
Title RegulatoryLandscape and Updates
Objective
Outline the framework of applicable regulatory guidelines and any recent
updates impacting validation.
Sources
List of reference documents from:– FDA (e.g., 21 CFR Part 11, Part
210/211)– EU Annex 15– ICH Guidelines (e.g., Q8, Q9, Q10)– WHO,
PIC/S, ISPE, GAMP 5– Local health authorities
Recent Changes
Summary of recent or significant regulatory changes (e.g., new FDA
guidance on Computer Software Assurance, updated EU GMP Annexes)
Impact on
Validation
Describe how updates affect:– Validation lifecycle– Documentation– Risk
assessment– Frequency of revalidation– Data integrity requirements
Action Plan
List of changes made or planned in the VMP or validation strategy to
address these updates.
What to Include in the Regulatory Updates Section
61.
Case Study: FDA483 Due to Poor Validation
Company Profile:
•Type: Pharmaceutical Manufacturer (Sterile
injectables)
•Location: U.S.-based facility
•Inspection Year: 2022
•Regulatory Body: U.S. FDA
Observation from FDA 483:
"Your firm failed to adequately validate the
sterilization process used for drug product
manufacturing. Specifically, your media fill
simulations lacked proper documentation,
acceptance criteria, and requalification
strategy."
Key Validation Failures Identified:
Area Issue
Process Validation
Media fill protocols did not match commercial manufacturing conditions.
No documented rationale for frequency or sampling plan.
Revalidation No periodic revalidation schedule for critical aseptic processes.
Data Integrity
Media fill results were missing batch records and lacked real-time entry
controls.
Change Control
Changes in sterilization cycle parameters were implemented without
impact assessment or revalidation.
Training
Operators conducting validation were not documented as trained on
revised SOPs.
62.
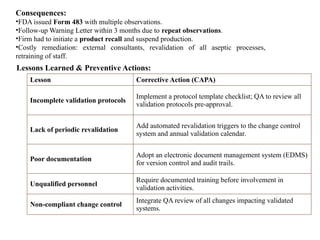
Consequences:
•FDA issued Form483 with multiple observations.
•Follow-up Warning Letter within 3 months due to repeat observations.
•Firm had to initiate a product recall and suspend production.
•Costly remediation: external consultants, revalidation of all aseptic processes,
retraining of staff.
Lessons Learned & Preventive Actions:
Lesson Corrective Action (CAPA)
Incomplete validation protocols
Implement a protocol template checklist; QA to review all
validation protocols pre-approval.
Lack of periodic revalidation
Add automated revalidation triggers to the change control
system and annual validation calendar.
Poor documentation
Adopt an electronic document management system (EDMS)
for version control and audit trails.
Unqualified personnel
Require documented training before involvement in
validation activities.
Non-compliant change control
Integrate QA review of all changes impacting validated
systems.
63.
How to ApplyThis in VMP:
•Include a section: "Case Studies and Industry Learnings"
•Add a risk mitigation strategy aligned with these learnings
•Tie in CAPAs with your validation SOPs, training programs, and change
control procedures
64.
Key Benefits ofa Robust VMP
Benefit Why It Matters
1. Regulatory
Compliance
Ensures all validation activities comply with FDA, EMA, WHO, MHRA, and
other regulatory bodies. Prevents 483s, warning letters, and product recalls.
2. Risk Management
Supports a systematic, risk-based approach (e.g., ICH Q9, GAMP 5) that
focuses resources on critical systems and processes.
3. Consistency &
Standardization
Establishes uniform procedures and documentation practices across all
departments and facilities.
4. Improved Audit
Readiness
Demonstrates control and traceability of validation lifecycle, making
inspections more manageable and less stressful.
5. Enhanced Quality
Assurance
Provides evidence that equipment, systems, and processes consistently perform
as intended, protecting product quality and patient safety.
6. Efficient Change
Control
Integrates validation into the change management process, ensuring
revalidation when needed and preventing unintended compliance gaps.
7. Better Resource
Planning
Allows scheduling of validation activities, avoiding overlaps, downtime, and
resource conflicts.
8. Clear Roles &
Responsibilities
Clearly defines team responsibilities, improving accountability and
communication.
9. Continuous
Improvement
Promotes regular review and updating of validation activities based on trends,
CAPAs, and regulatory changes.
10. Cost Reduction
Over Time
Prevents expensive remediation, product failures, and rework by getting
validation right the first time.
65.
Conclusion
The Validation MasterPlan (VMP) is a critical, high-level document that outlines a company's
overall strategy and approach to validation across systems, equipment, facilities, utilities,
processes, and computer systems. A well-structured VMP ensures that all validation activities are
planned, executed, and documented in a consistent, risk-based, and compliant manner.
Key Takeaways:
• The VMP serves as a roadmap for ensuring that systems and processes meet regulatory
requirements (e.g., FDA, EMA, WHO).
• It defines validation objectives, scope, responsibilities, documentation requirements, revalidation
triggers, and risk management strategies.
• Proper implementation of the VMP ensures data integrity, product quality, and patient safety.
• Integration of GAMP 5, ICH guidelines, and change control mechanisms enhances the robustness
of the validation framework.
• Regular review and updates of the VMP keep the organization aligned with current regulatory
expectations and industry best practices.
“A robust VMP is not just a regulatory requirement — it is a cornerstone of a
company’s quality and compliance strategy.”
66.
Thank You!
Thank youfor your attention and commitment to quality and compliance.
A well-executed VMP not only fulfills regulatory requirements but lays the
foundation for operational excellence and patient safety.
"Validation is not a one-time task — it’s a culture of accountability.
A strong VMP transforms compliance from a checkbox into a commitment."
— Quality Assurance Principle