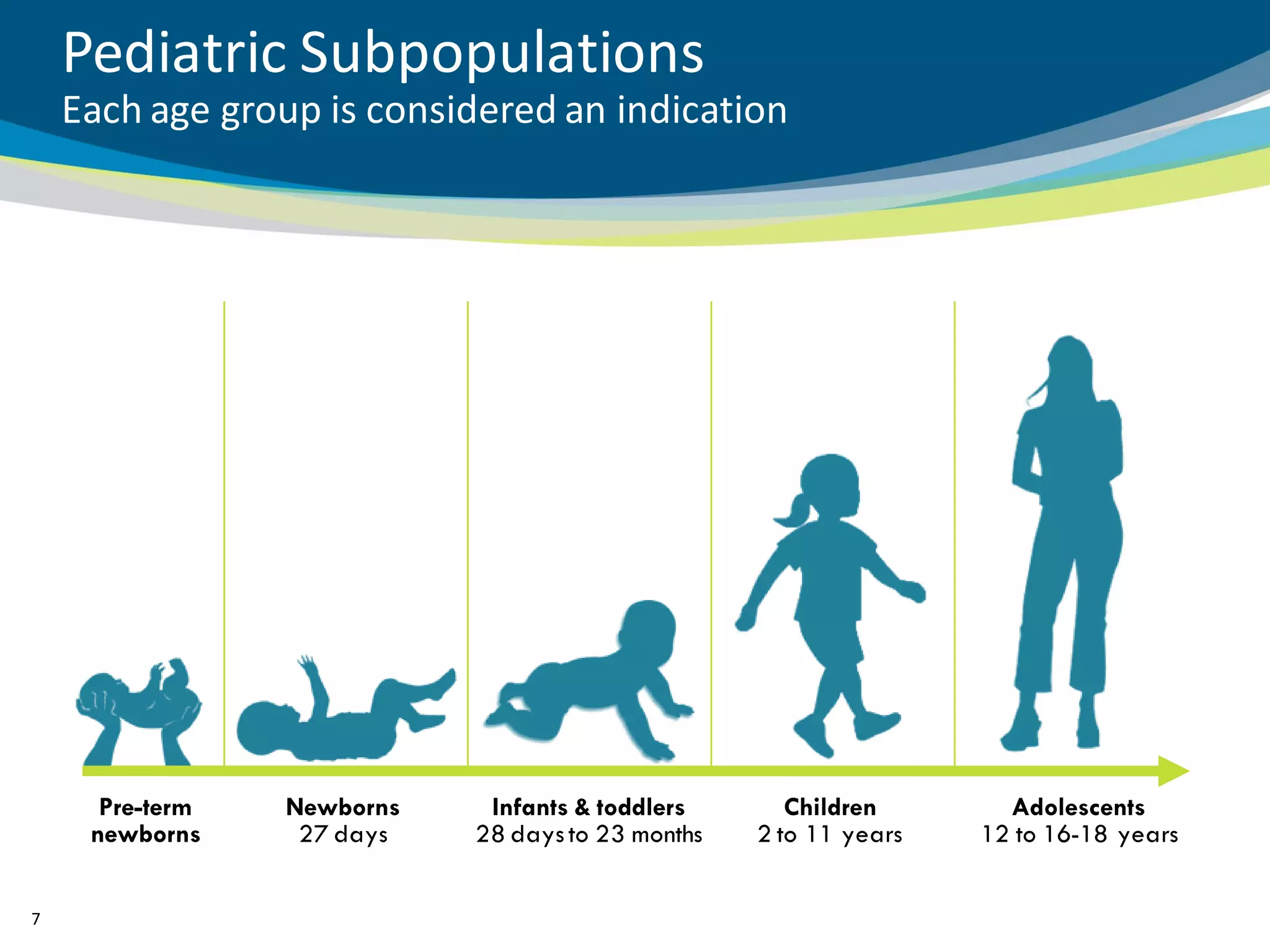
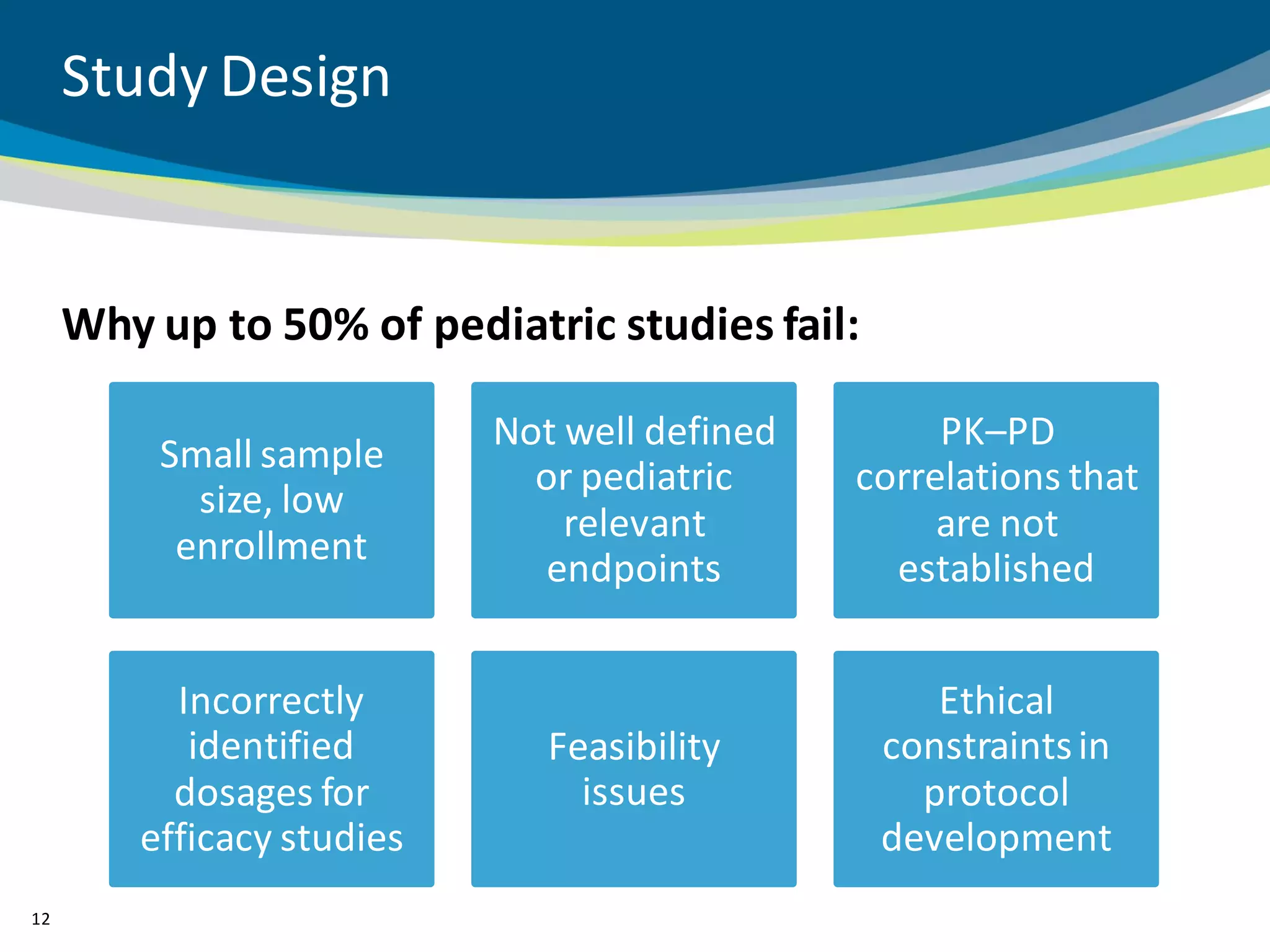
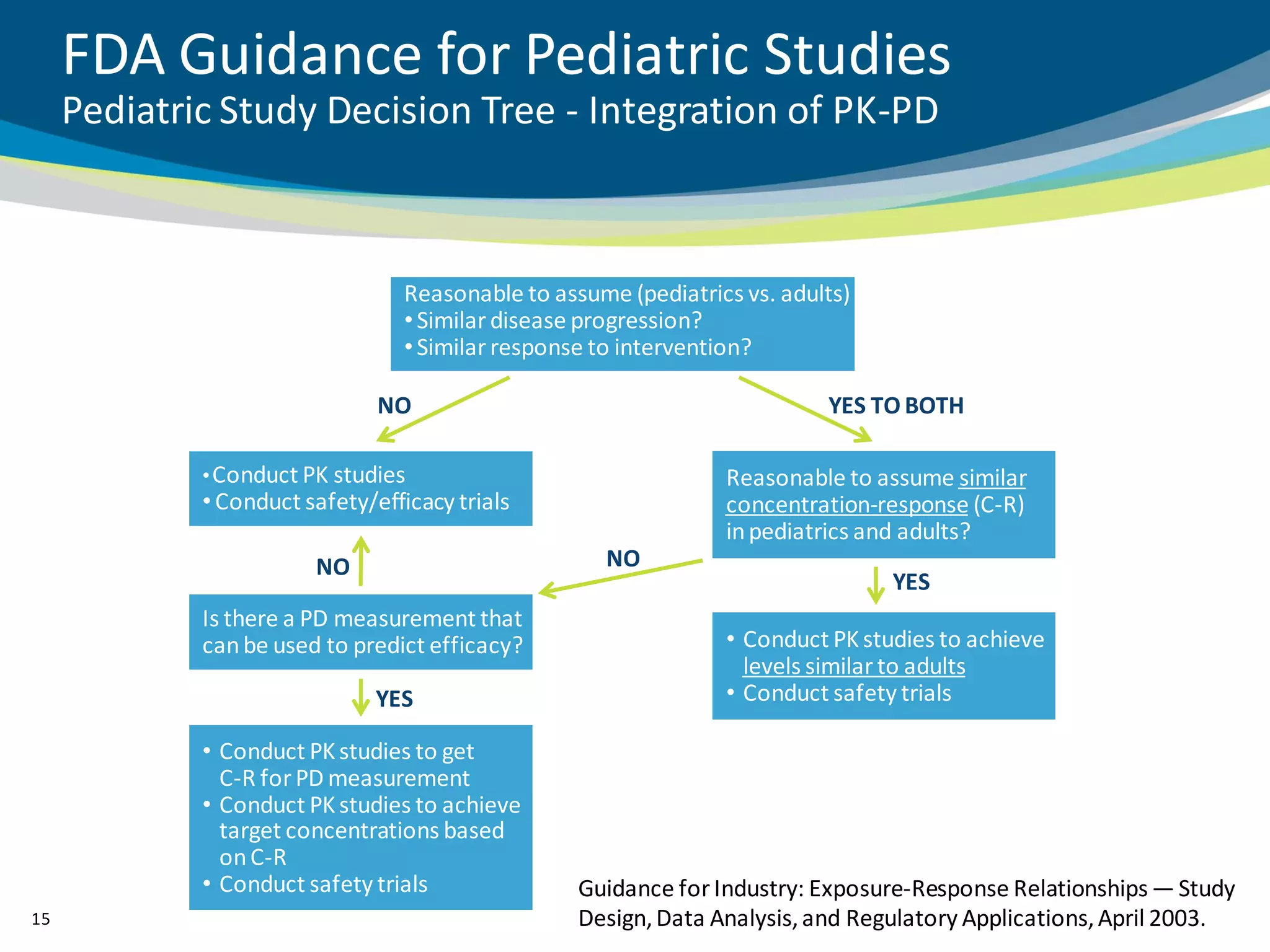
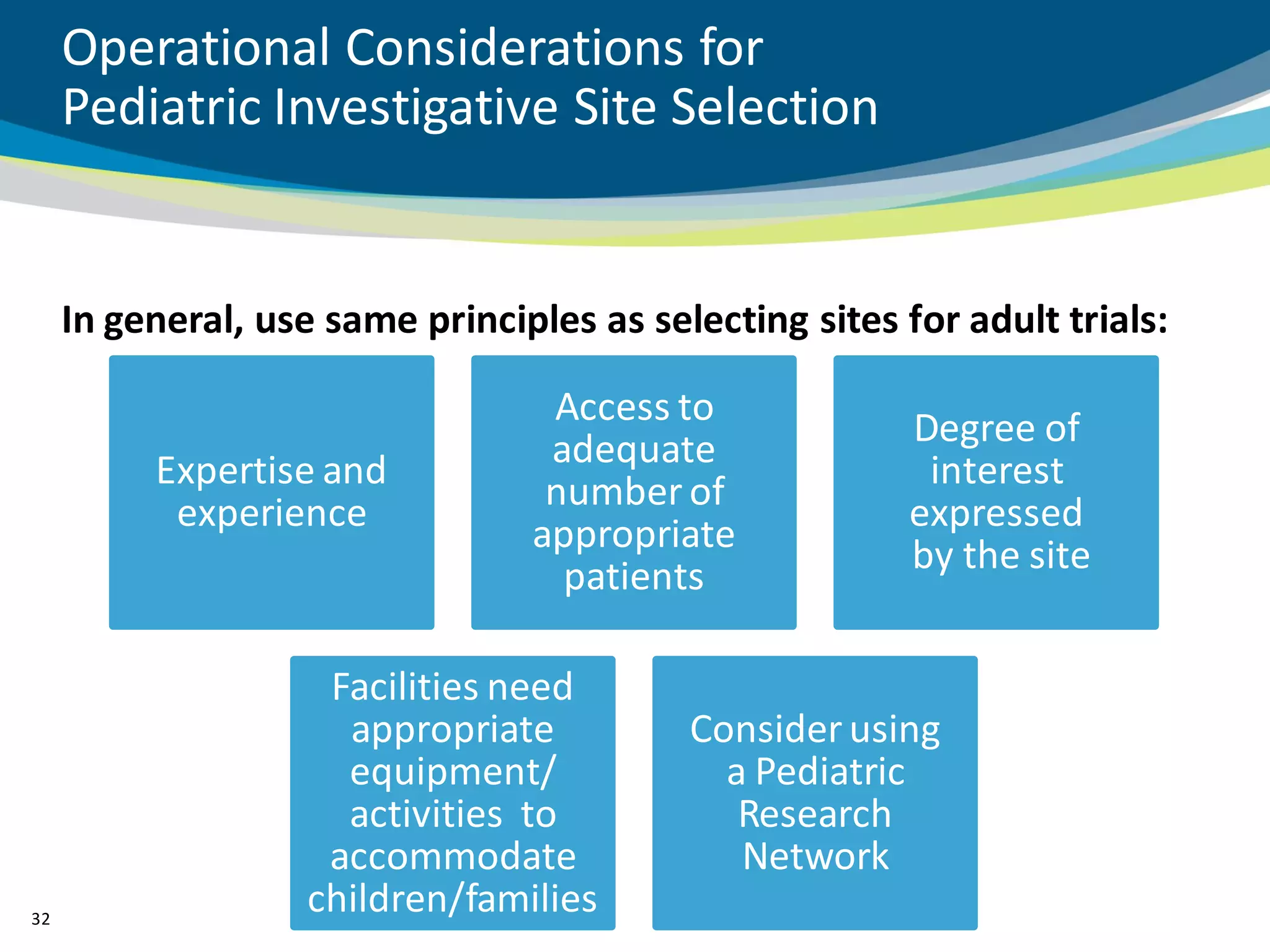
The document discusses key considerations for designing successful pediatric studies, highlighting the differences between pediatric and adult research protocols. It emphasizes the need for age-appropriate formulations, ethical protections for vulnerable populations, and the importance of engaging families in the research process to enhance enrollment and retention. Additionally, it outlines various recruitment strategies and operational considerations specific to pediatric participants to ensure effective study implementation.