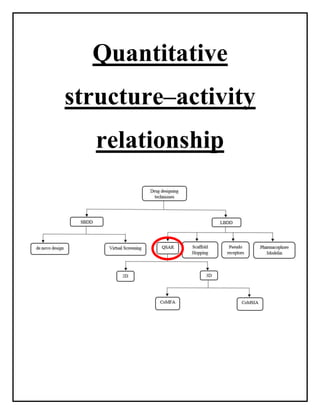
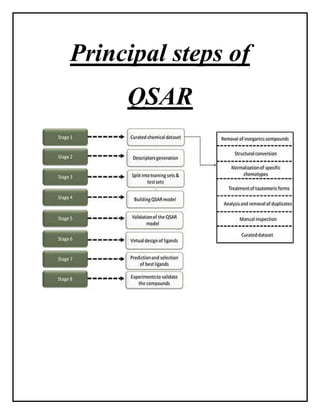
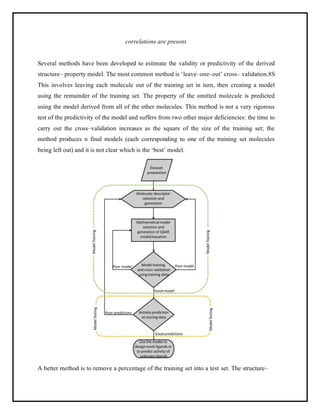
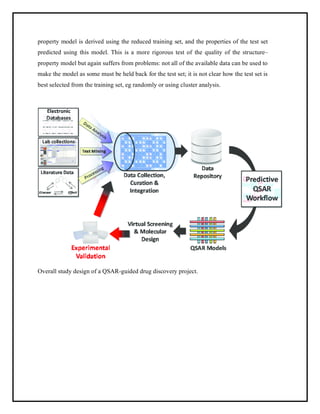
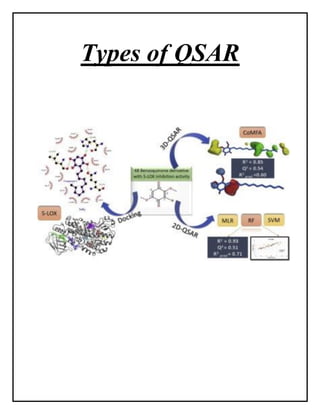
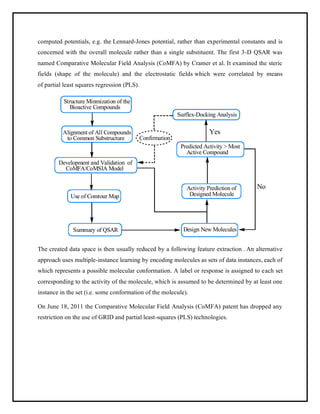
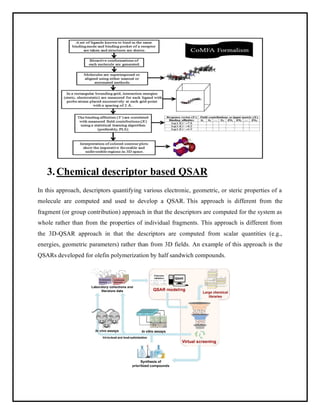
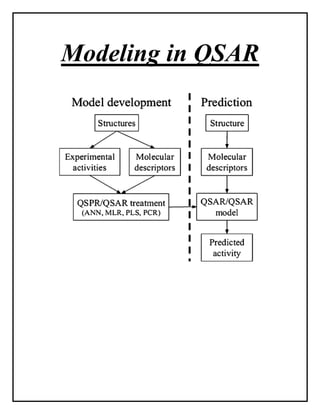
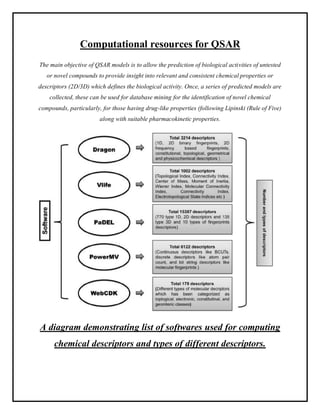
This document discusses Quantitative Structure-Activity Relationship (QSAR) modeling. It provides an introduction to QSAR, outlines the basic mathematical form of QSAR models, and describes several key steps in the QSAR modeling process including generating molecular descriptors, selecting descriptors, mapping descriptors to biological activities, and validating models. The principal steps are described as selection of data and descriptors, variable selection, model construction, and validation/evaluation. Various types of molecular descriptors are also defined including topological, geometric, electronic, and hybrid descriptors.