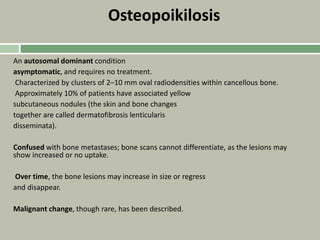
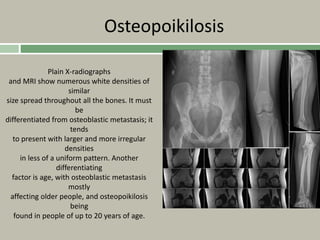
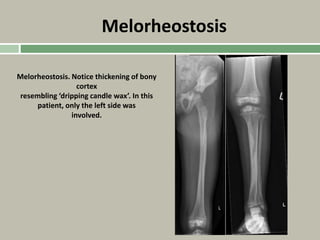
Downloaded 19 times







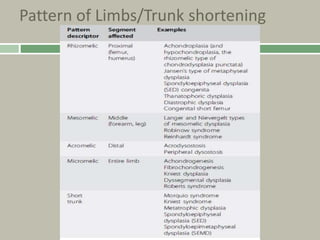

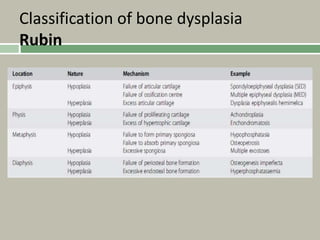







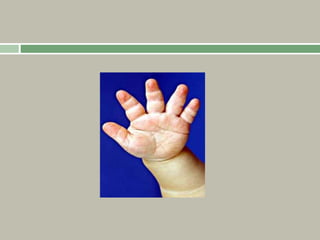


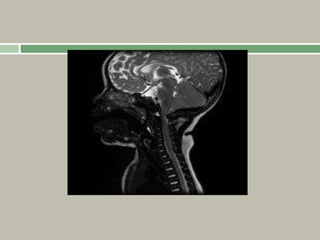
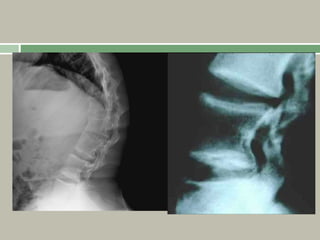
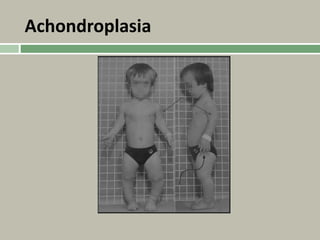



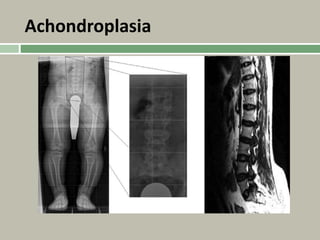












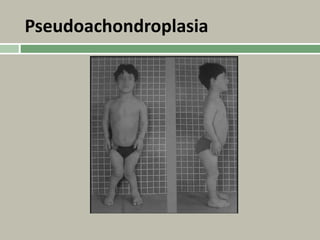
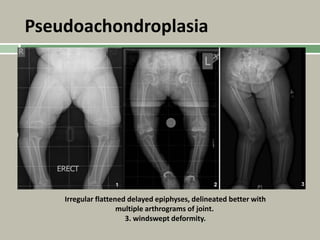













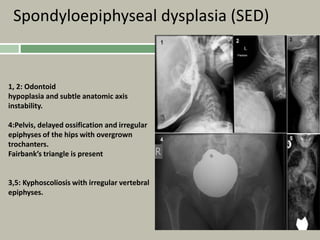
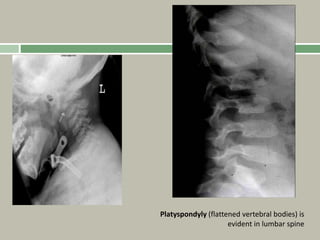









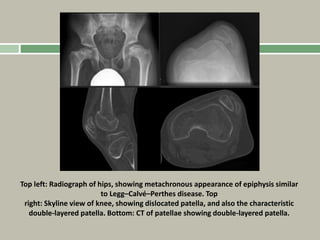
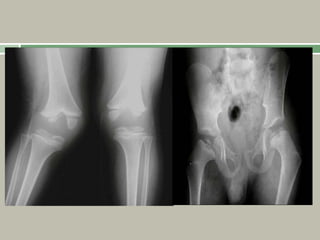








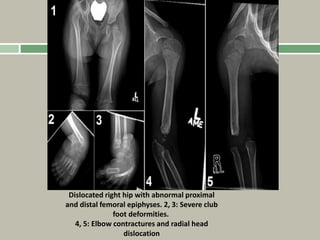
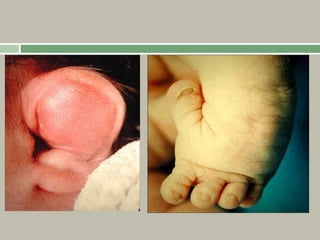
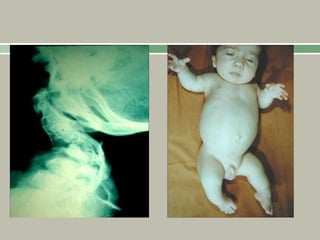



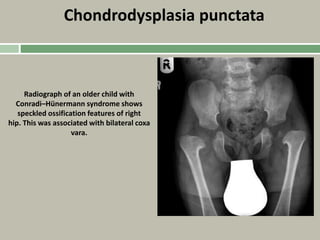






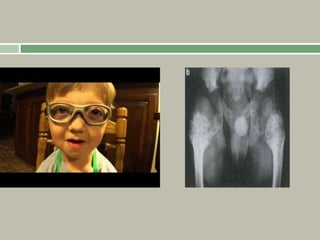
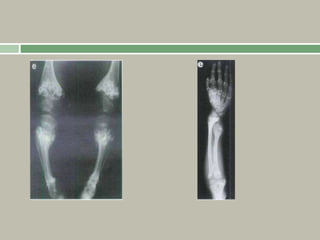

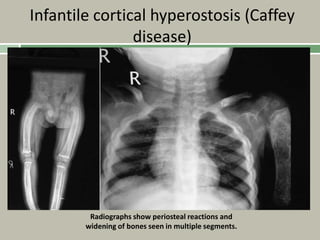




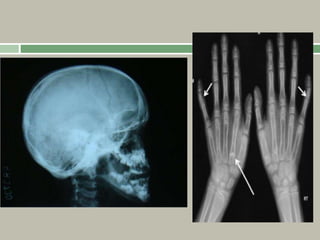
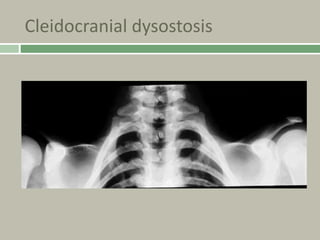
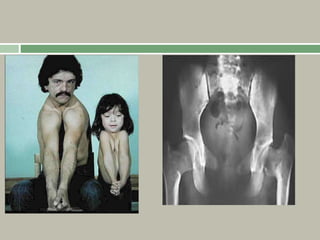




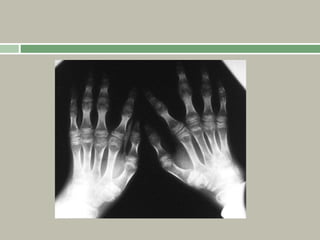






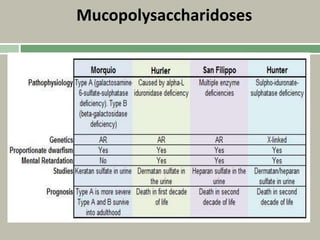


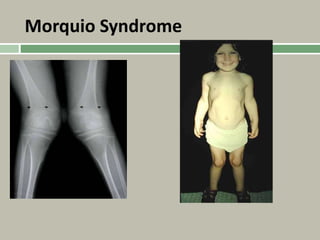

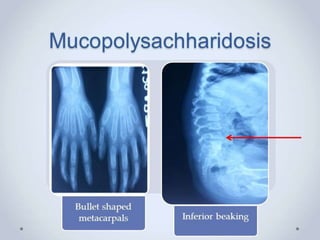




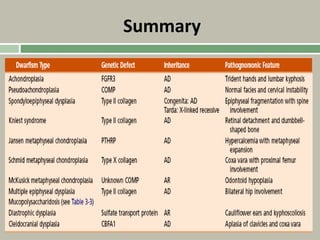

1. A skeletal dysplasia is a congenital abnormality of bone growth or development that results in structural abnormalities of the bones. 2. Making a diagnosis involves taking a thorough history and physical examination, including measurements of height, limb lengths, and facial features. Radiographs can identify which bones are affected. 3. Achondroplasia is the most common skeletal dysplasia, caused by a mutation in the FGFR3 gene, and is characterized by disproportionate short stature, frontal bossing, trident hands, genu varum, and foramen magnum stenosis.























































































