Downloaded 152 times


















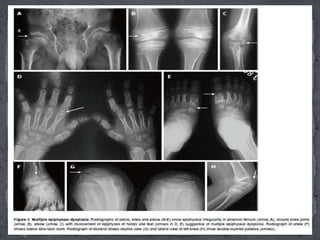







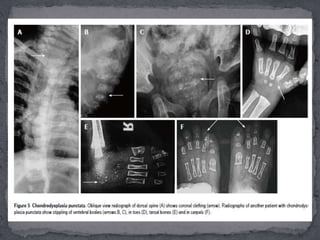



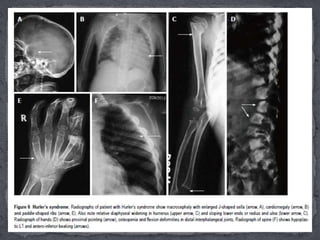


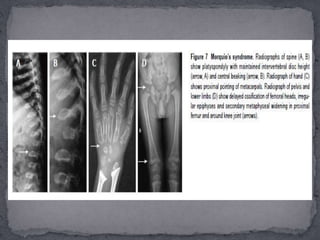



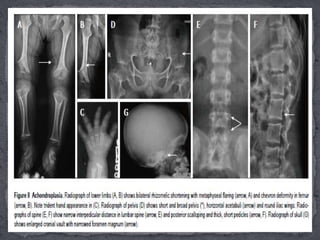


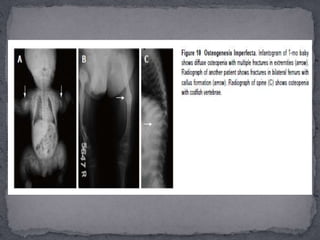



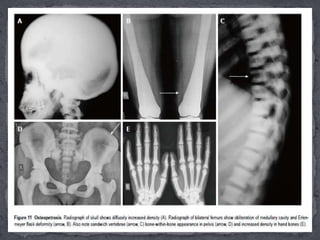






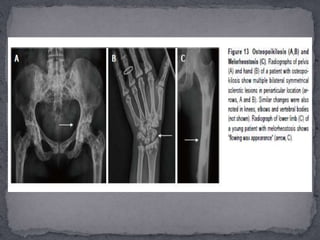





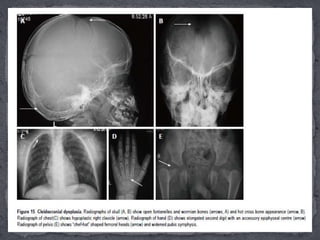
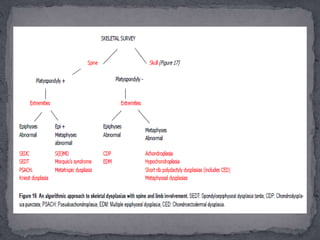
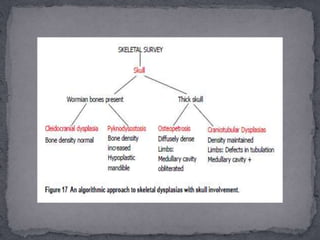



This document discusses skeletal dysplasias (osteochondrodysplasias), which are disorders involving abnormalities of bone or cartilage growth and texture. It describes the classification of skeletal dysplasias into four groups based on their anatomical location and characteristics. Key radiographic features of several common skeletal dysplasias are outlined, including spondyloepiphyseal dysplasia congenita (SEDC), spondyloepiphyseal dysplasia tarda (SEDT), multiple epiphyseal dysplasia (MED), pseudoachondroplasia, chondrodysplasia punctata (CDP), mucopolysaccharidoses (MPS), Morquio syndrome


















































































