







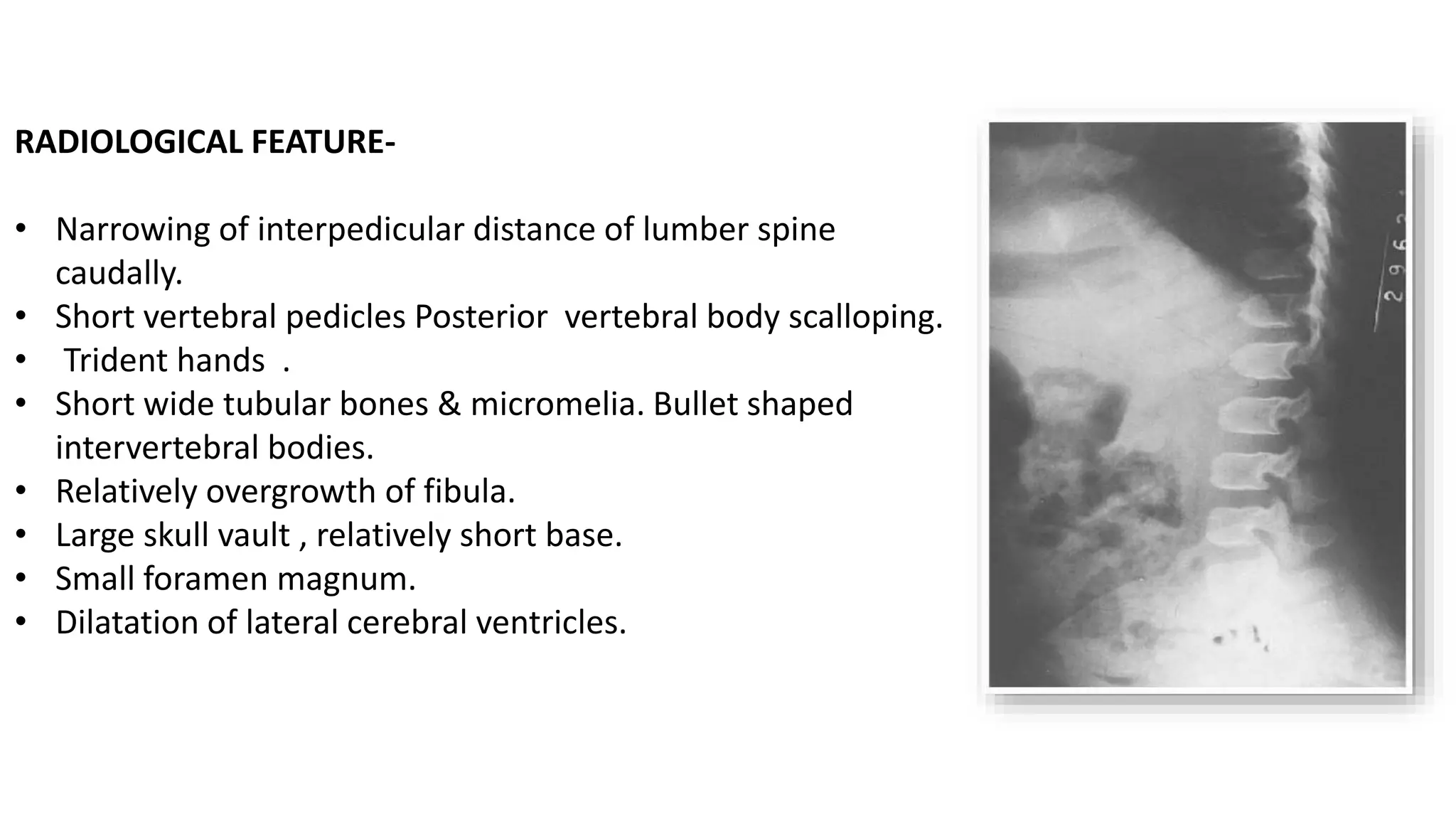






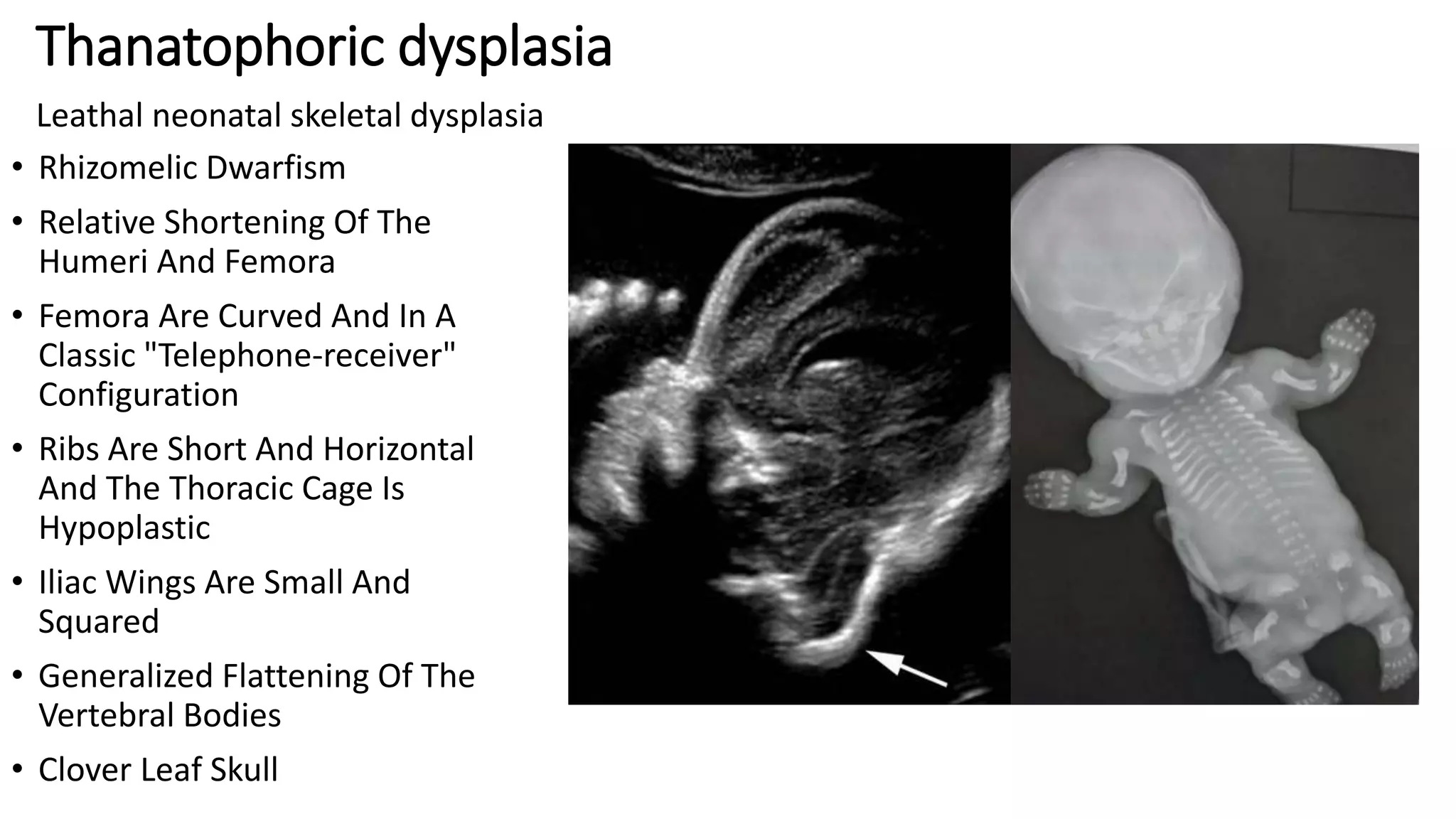




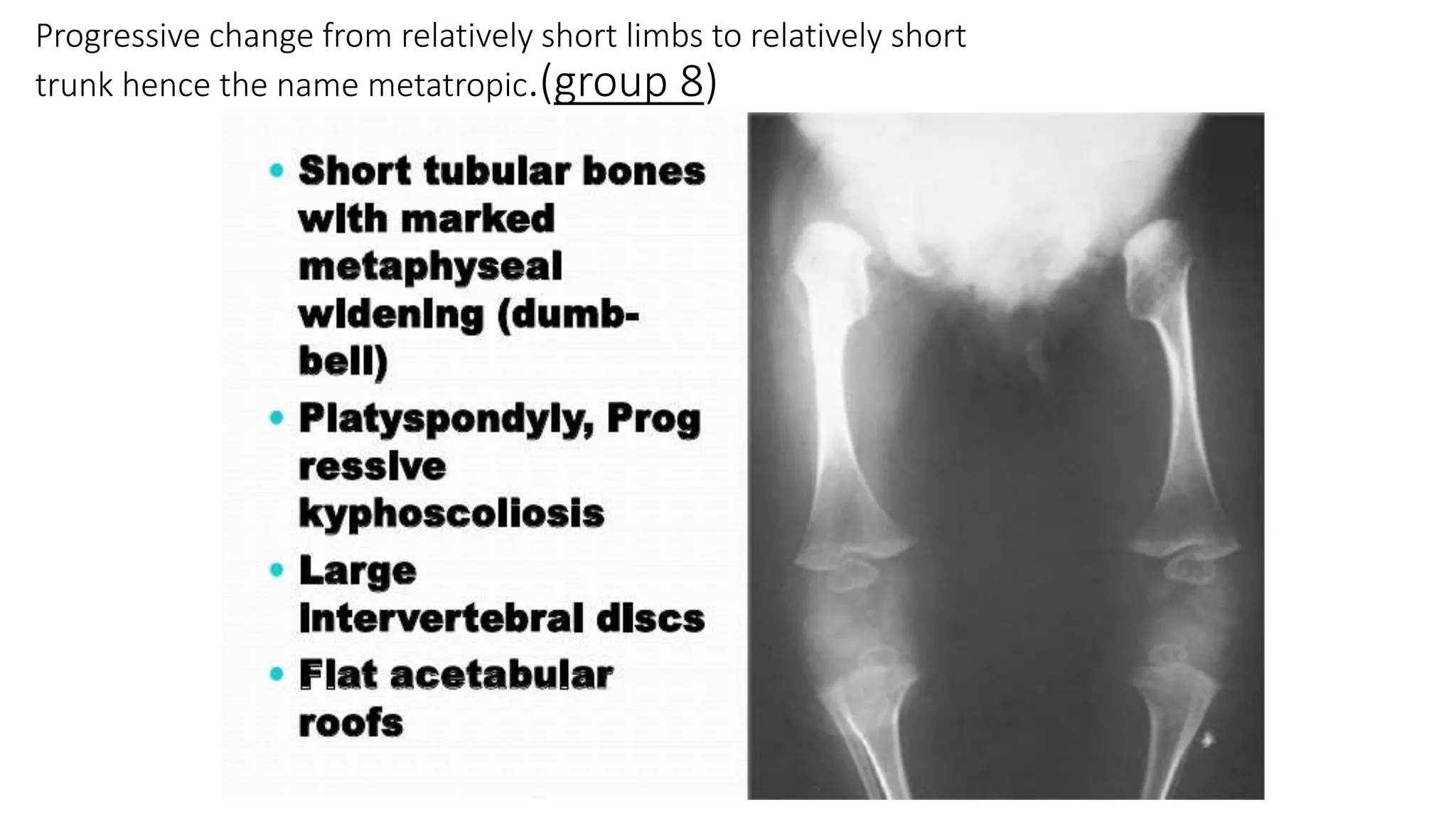







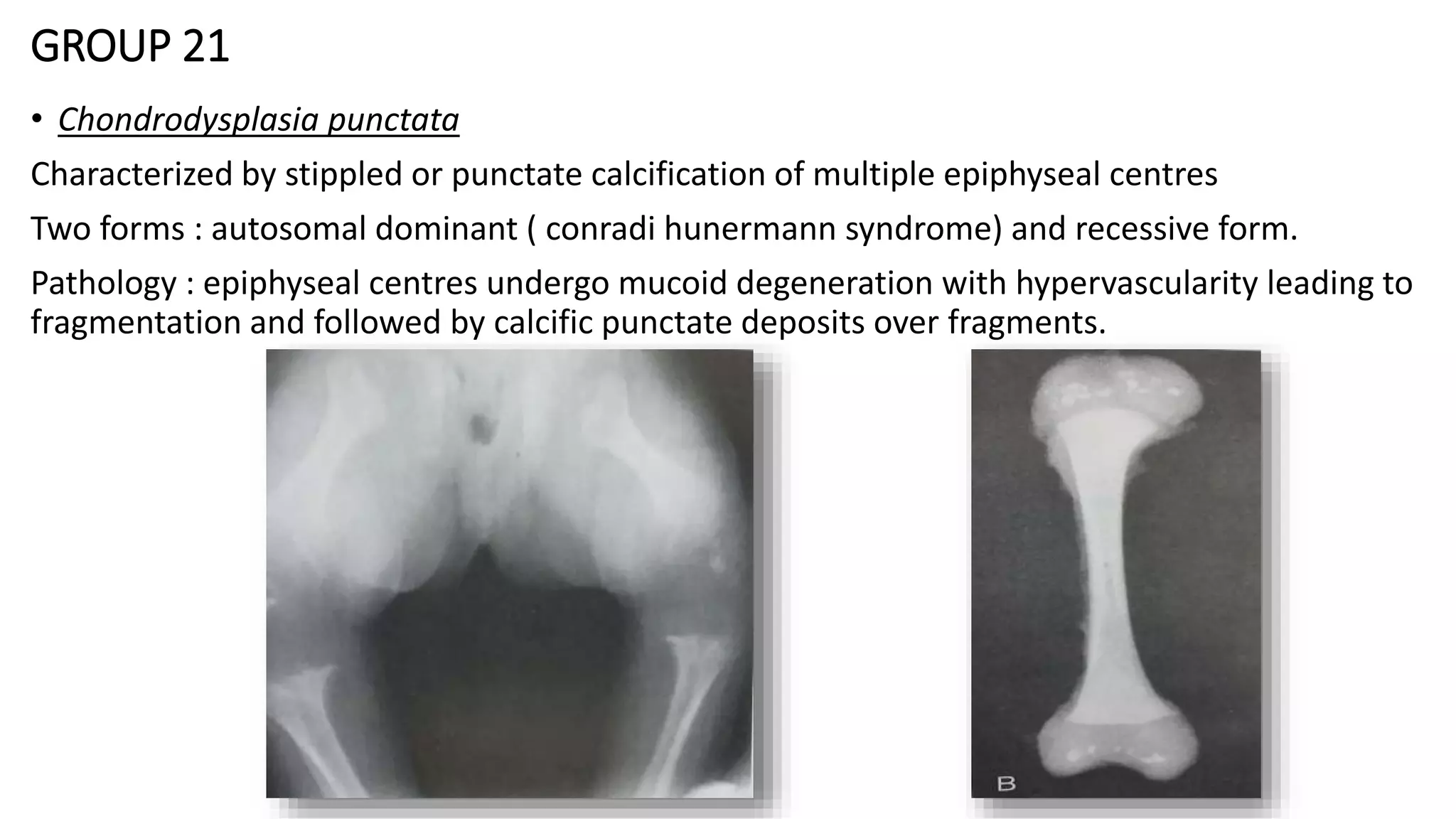





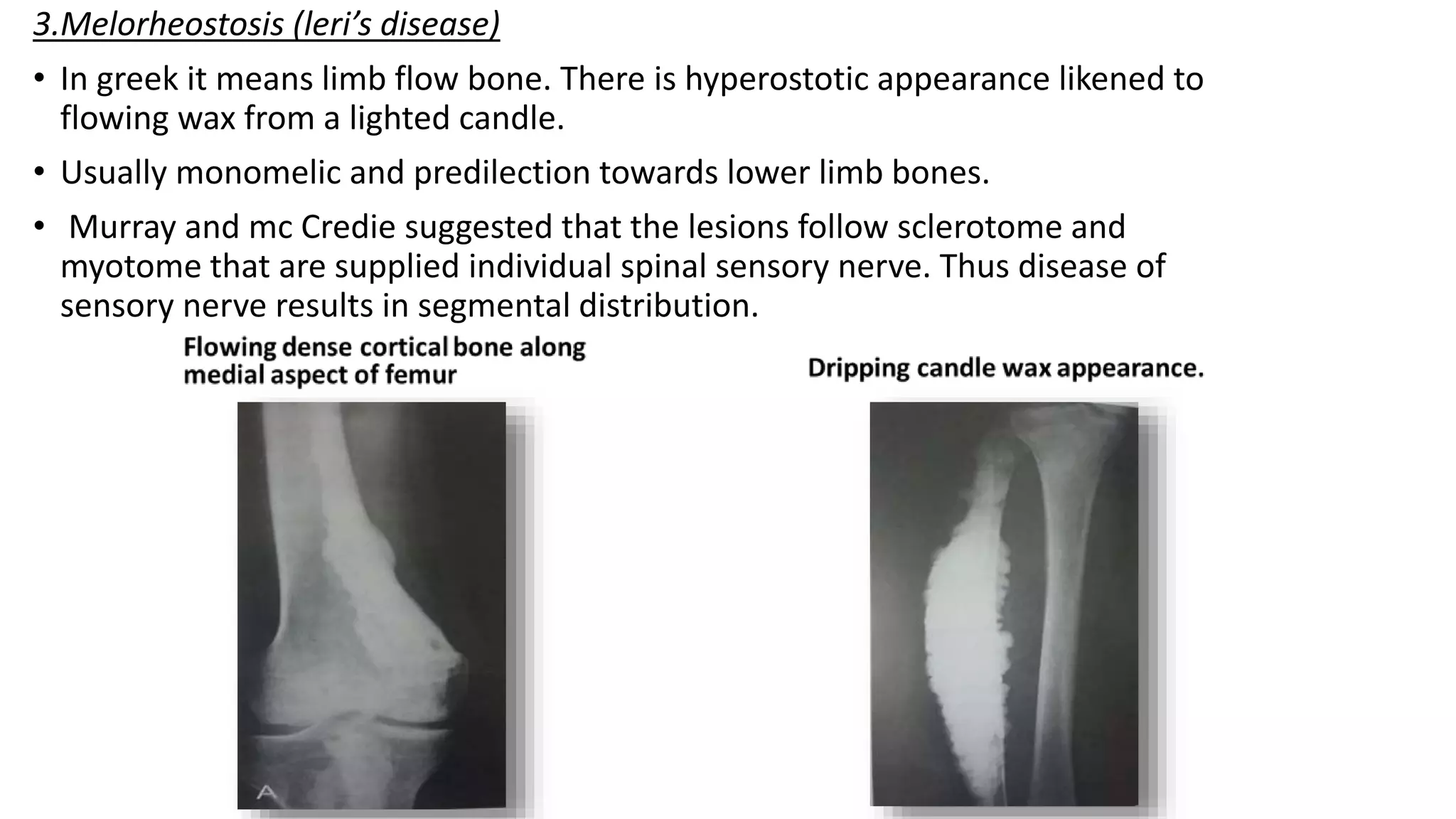





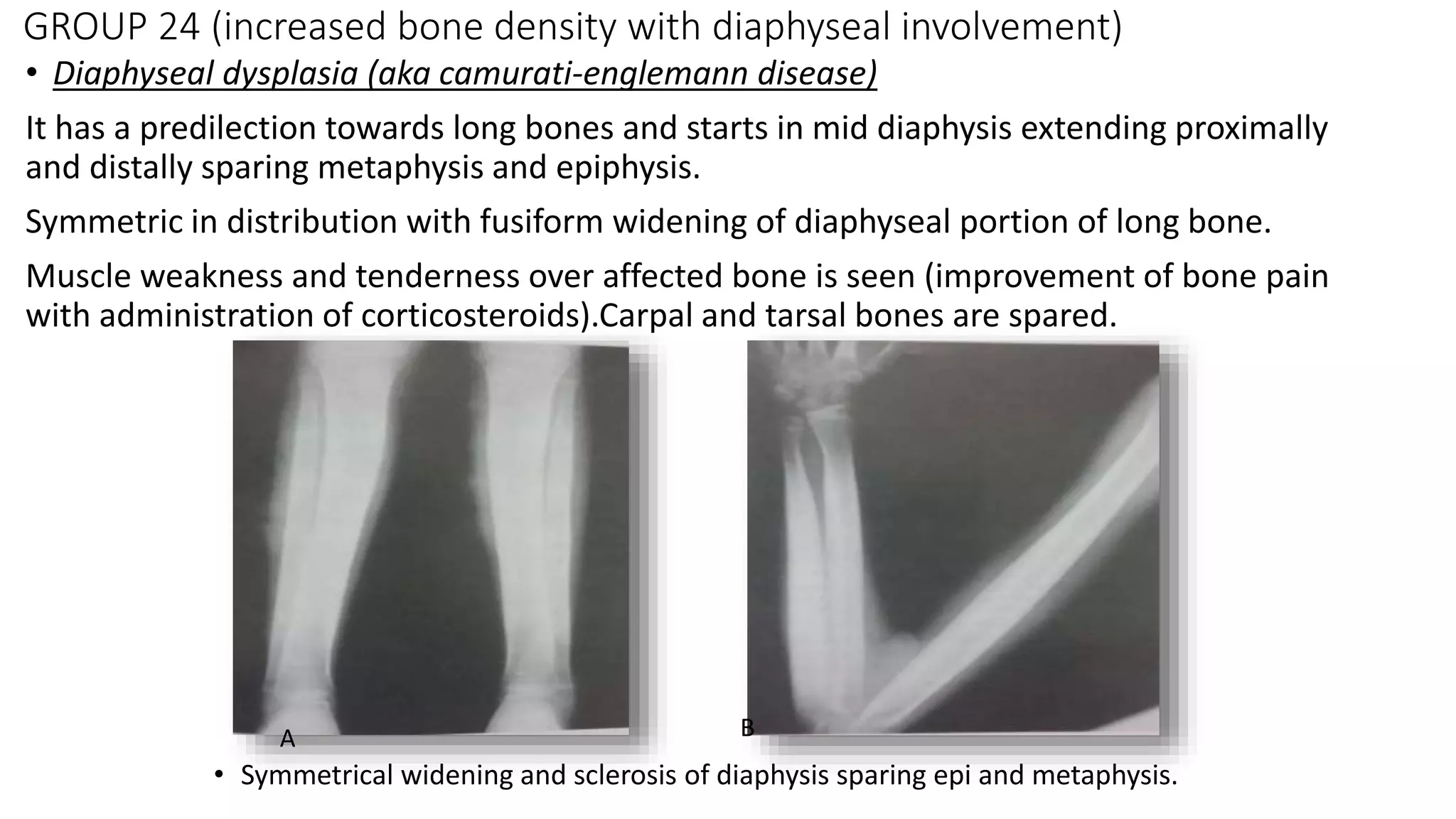




































This document summarizes imaging findings in skeletal dysplasias. It discusses over 20 different conditions classified into groups based on genetic and phenotypic characteristics. Key radiological features are provided for common dysplasias like achondroplasia and spondyloepiphyseal dysplasia. A skeletal survey approach is outlined to identify clues for diagnosis. Prenatal imaging and multidisciplinary evaluation are important for diagnosis and genetic counseling.














































































