Downloaded 170 times

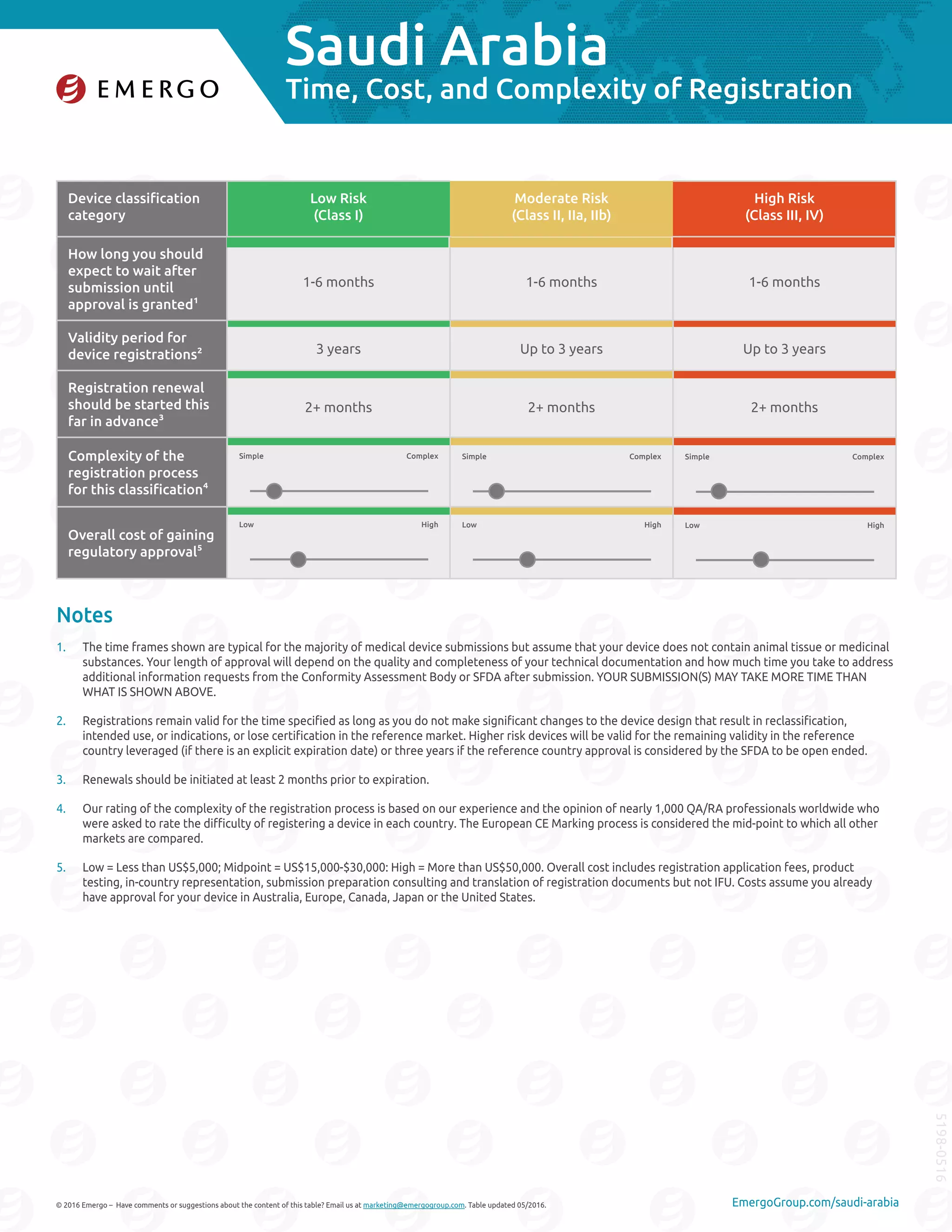

To market medical devices in Saudi Arabia, devices must receive marketing authorization from the Saudi Food and Drug Authority (SFDA). The SFDA application is reviewed for completeness then sent to a third party for technical review, which can involve multiple rounds of questions. Once approved, the SFDA issues a certificate allowing marketing in Saudi Arabia. Appointing an authorized local representative is also required to manage the registration process. Overall, registration times range from 1-6 months but costs and complexity vary depending on the device's risk classification.









































































