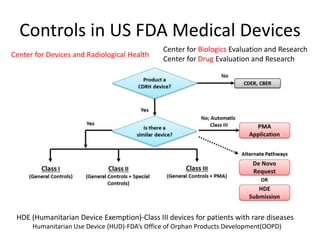
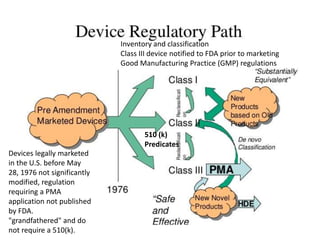
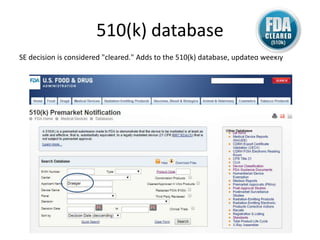
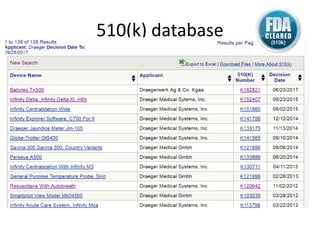
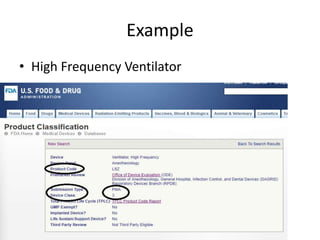
The US FDA regulates medical devices to ensure they are safe and effective. There are three classes of medical devices with increasing regulatory controls based on risk. Class I devices have general controls, Class II devices have general and special controls, and Class III devices require premarket approval. To market a device in the US, manufacturers must classify their device, submit the appropriate premarket notification or application, and complete an establishment registration and device listing. The FDA reviews submissions to ensure devices reasonably assure safety and effectiveness.















































![Sec. 807.100 FDA action on a
premarket notification.
(b) FDA will determine that a device is substantially equivalent to a predicate device
using the following criteria:
(1) The device has the same intended use as the predicate device; and
(2) The device:
(i) Has the same technological characteristics as the predicate device; or
(ii)(A) Has different technological characteristics, such as a significant change in the
materials, design, energy source, or other features of the device from those of the
predicate device;
(B) The data submitted establishes that the device is substantially equivalent to the
predicate device and contains information, including clinical data if deemed
necessary by the Commissioner, that demonstrates that the device is as safe and
as effective as a legally marketed device; and
(C) Does not raise different questions of safety and effectiveness than the predicate
device.
(3) The predicate device has not been removed from the market at the initiative of
the Commissioner of Food and Drugs or has not been determined to be
misbranded or adulterated by a judicial order.
[57 FR 58403, Dec. 10, 1992, as amended at 63 FR 5253, Feb. 2, 1998]](https://image.slidesharecdn.com/usfda-191214045417/85/US-FDA-Medical-Device-or-Equipment-63-320.jpg)












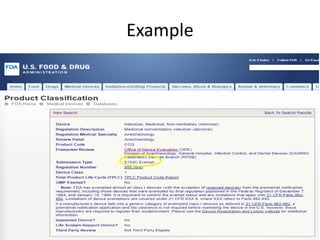
![Steps in the PMA Application Process
• Substantive review coordination and completion in areas such as:
– Preparation of FDA Summary of Safety and Effectiveness Data (SSED)
– Nonclinical Studies
[Microbiological, Toxicological, Immunological, Biocompatibility, Shelf
Life, Analytical (for IVDs), Animal, Engineering (Stress, Wear, Fatigue,
etc.)]
– Clinical Studies
– Panel Meeting Decision and Mailing (if panel meeting is appropriate)
– Panel Date (if appropriate)
– Transcripts Received, Reviewed and Placed in Administrative Record
– QS/GMP Clearance
– Final Response from OC for GMP/BIMO
– Final ODE Decision Memo
– Approval Package
– Approval Order, SSED, Final Draft Labeling](https://image.slidesharecdn.com/usfda-191214045417/85/US-FDA-Medical-Device-or-Equipment-78-320.jpg)