Downloaded 717 times
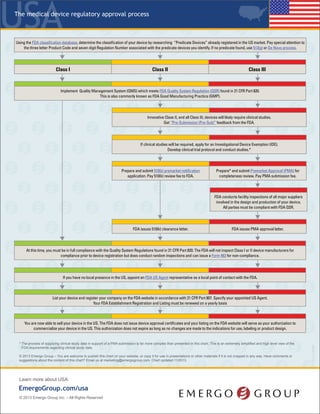

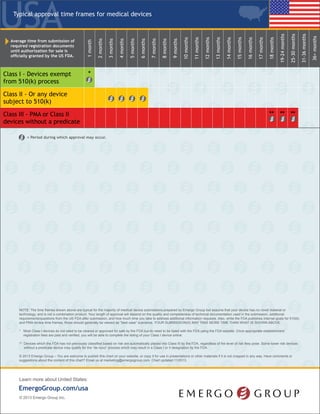

The document outlines the regulatory process for classifying and registering medical devices with the FDA in the United States, including details on device classification, necessary documentation, associated fees, and timelines for approval. Class I devices generally do not require FDA premarket clearance, whereas Class II and Class III devices usually necessitate clinical studies and more extensive submissions. It emphasizes the importance of compliance with quality management standards and the need for annual registration renewals to maintain market authorization.



































































