








![ When manufacturing pharmaceuticals in Japan, each
manufacturing location requires a manufacturing license.
As a condition to receiving a manufacturing license, there
must be a full-time pharmacologist in each manufacturing
location who serves as the Pharmaceuticals Manufacturing
Manager (Managing Pharmacist).]
The manufacturing license is issued by the governor of the
prefecture in which the manufacturing location is located
except in cases requiring particularly high levels of
expertise.](https://image.slidesharecdn.com/medicaldeviceapprovalprocessinjapanppt-111213035407-phpapp01/75/MEDICAL-DEVICE-APPROVAL-PROCESS-IN-JAPAN-10-2048.jpg)







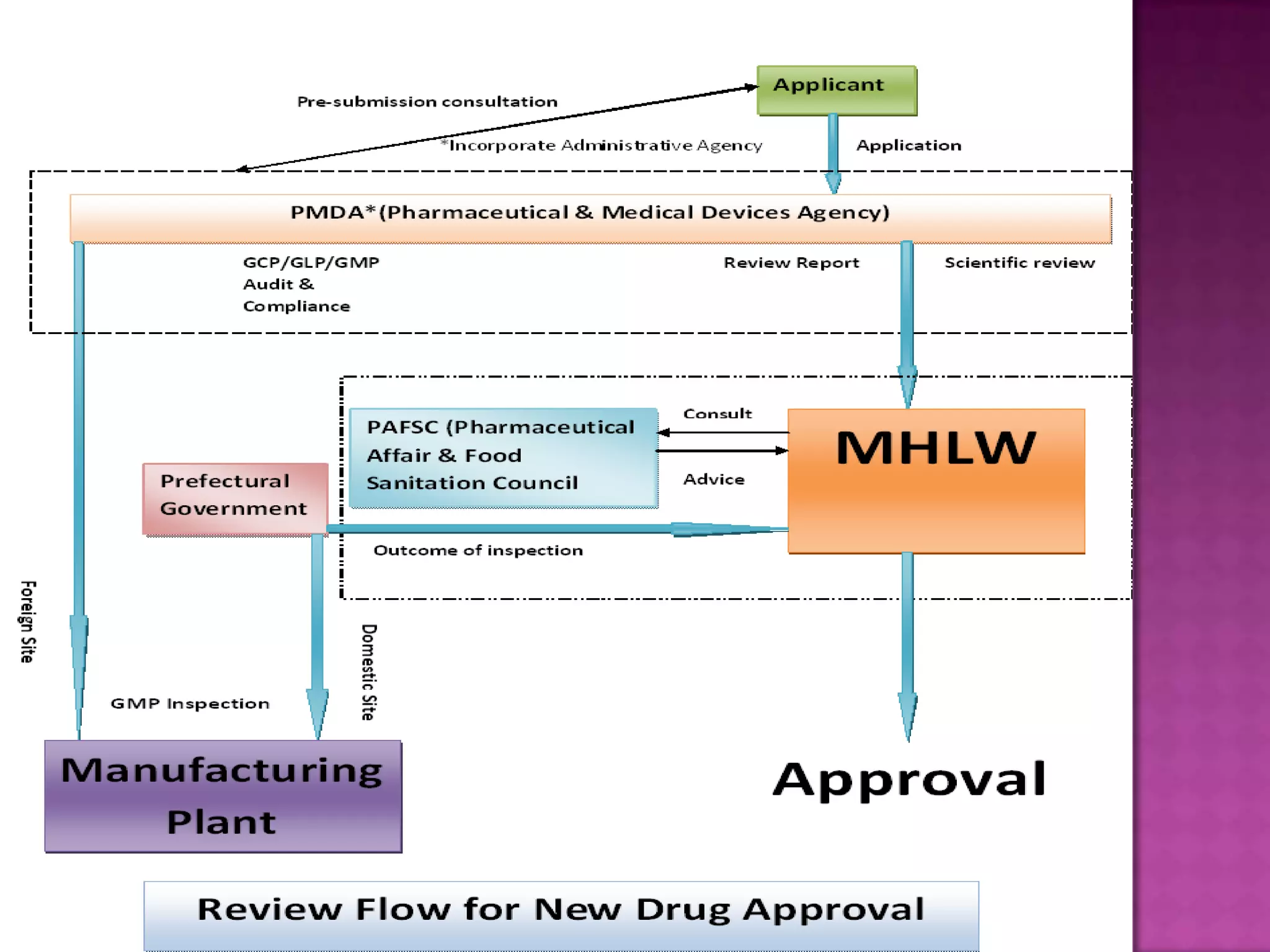
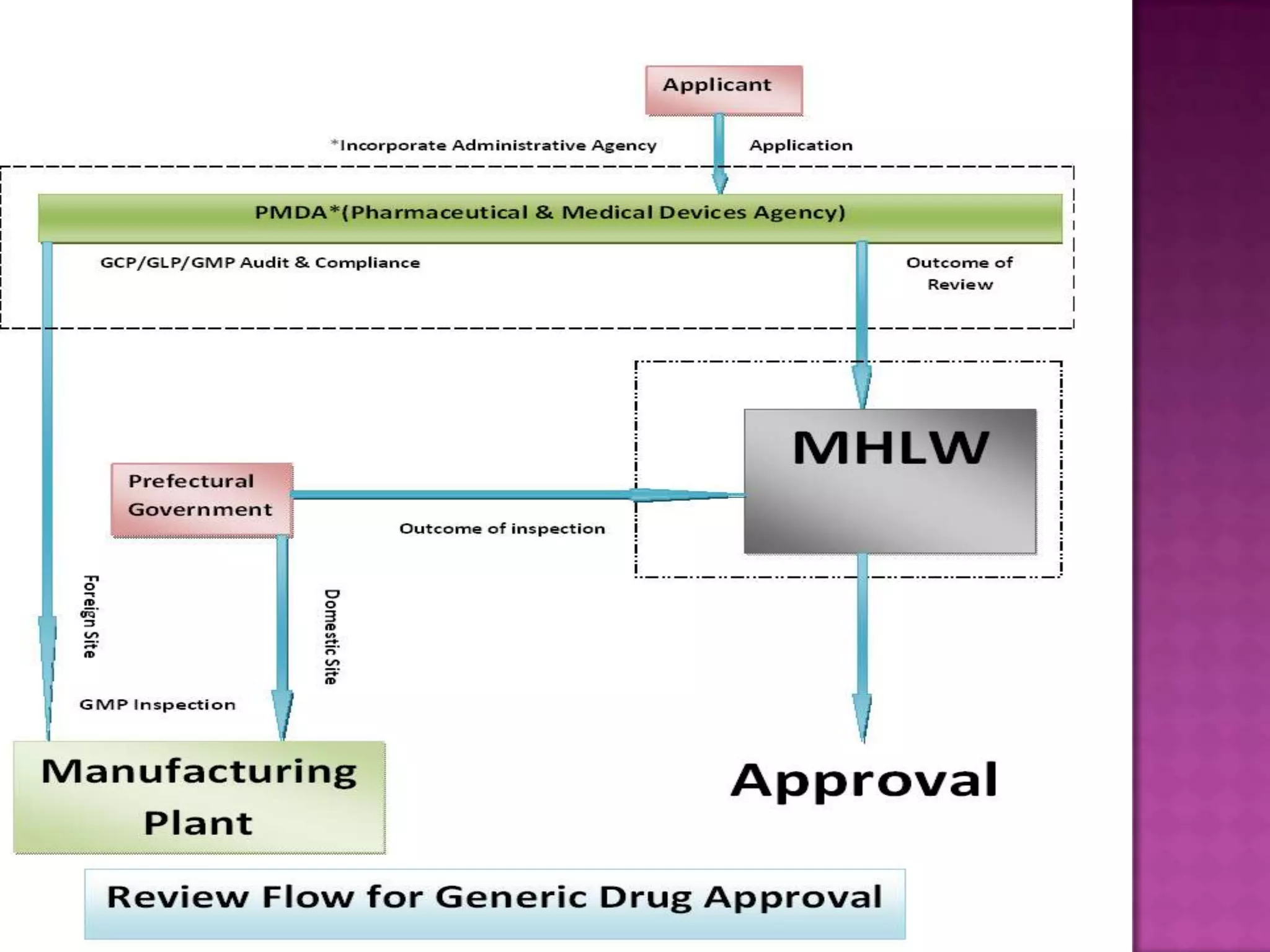

































The document discusses pharmaceutical regulations in Japan. It notes that Japan has the second largest pharmaceutical market behind the US. It also discusses regulations for clinical trials, marketing approval, and post-marketing safety measures. The key regulatory bodies are the Ministry of Health, Labor and Welfare (MHLW) and the Pharmaceuticals and Medical Devices Agency (PMDA). Pharmaceutical companies must follow good manufacturing practice (GMP) standards and obtain various approvals to market and sell drugs in Japan.




















![European_Union.ppt.Nikhil[1].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/europeanunion-220803170320-4be1aa31-thumbnail.jpg?width=640&height=640&fit=bounds)


































































