Downloaded 34 times







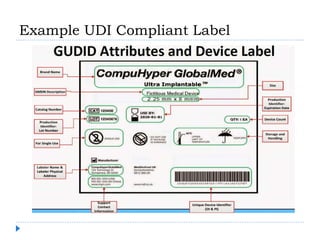








The document outlines the history and implementation of the Unique Device Identification (UDI) system for medical devices, initiated by the FDA under the FDA Safety and Innovation Act. It details regulatory milestones, the final rule for UDI labeling, and the establishment of the Global UDI Database (GUDID) to improve safety and tracking of medical devices. Key benefits of UDI implementation include enhanced adverse event reporting, reduction in medical errors, and streamlined device recalls.

























![European_Union.ppt.Nikhil[1].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/europeanunion-220803170320-4be1aa31-thumbnail.jpg?width=640&height=640&fit=bounds)










































![ONFH[AVN HIP] -TRIPLE REGIME -A NOVAL SURGICAL CONCEPT .pptx](https://cdn.slidesharecdn.com/ss_thumbnails/onfhavnhip2026koaconcalicutdrgokuldevdrmashraf-260210064517-213ec005-thumbnail.jpg?width=640&height=640&fit=bounds)
















