Downloaded 827 times








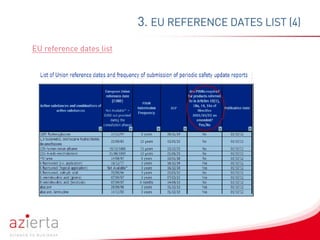




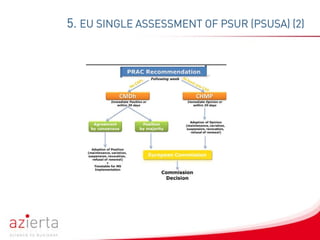





Periodic Safety Update Reports (PSURs) are pharmacovigilance documents evaluating the risk-benefit balance of medicinal products post-authorisation. The reports are submitted at defined intervals and must incorporate new information on safety and efficacy, with updates occurring periodically based on regulatory guidelines. Exemptions exist for specific product categories, but marketing authorization holders are expected to continuously assess safety data even if PSUR submissions are not required.







































![Volume 9 A Guidelines On Pharmacovigilance[1]](https://cdn.slidesharecdn.com/ss_thumbnails/volume9aguidelinesonpharmacovigilance1-12816046179281-phpapp02-thumbnail.jpg?width=640&height=640&fit=bounds)









































