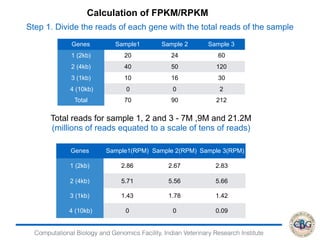
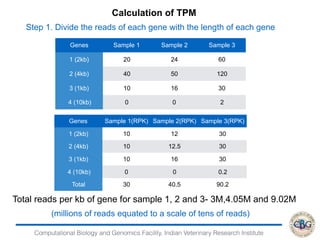
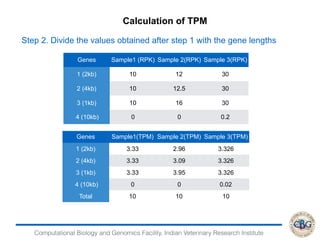
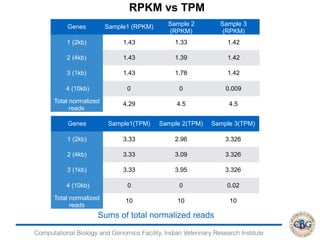
This document discusses various methods for normalization of RNA-seq read count data, including RPKM/FPKM, TMM, Limma voom, and TPM. It provides explanations of each method and how they aim to correct for differences in sequencing depth, transcript length, and transcript pool composition between samples. The document also provides examples of calculating RPKM, TPM, and comparing the two methods. Lastly, it discusses using tools like RSEM, Bowtie, and EBSeq for determining differentially expressed genes from RNA-seq data through a count-based strategy.





























![Computational Biology and Genomics Facility, Indian Veterinary Research Institute
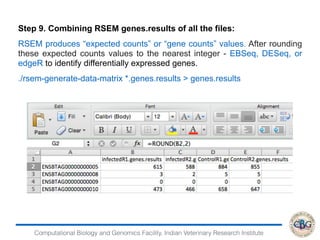
Step 6. Commands to Run EBSeq:
> x=data.matrix(read.table("genesresults.txt"))
> dim(x)
[1] 24596 4
> str(x)
num [1:24596, 1:4] 615 3 0 473 1 286 832 362 103 17 ...
- attr(*, "dimnames")=List of 2
..$ : chr [1:24596] "ENSBTAG00000000005" "ENSBTAG00000000008"
"ENSBTAG00000000009" "ENSBTAG00000000010" ...
..$ : chr [1:4] "infectedR1.genes.results" "infectedR2.genes.results"
"ControlR1.genes.results" "ControlR2.genes.results"
> Sizes=MedianNorm(x)
> EBOut=EBTest(Data=x,
+ Conditions=as.factor(rep(c("C1","C2"),each=2)),sizeFactors=Sizes,
+ maxround=5)](https://image.slidesharecdn.com/rsemdepackages-180111065110/85/RSEM-and-DE-packages-37-320.jpg)
![Computational Biology and Genomics Facility, Indian Veterinary Research Institute
Removing transcripts with 75 th quantile < = 10
12071 transcripts will be tested
iteration 1 done
time 0.12
iteration 2 done
time 0.13
iteration 3 done
time 0.08
iteration 4 done
> PP=GetPPMat(EBOut)
> str(PP)
num [1:12071, 1:2] 1 1 0 0 1 ...
- attr(*, "dimnames")=List of 2
..$ : chr [1:12071] "ENSBTAG00000000005"
"ENSBTAG00000000010" "ENSBTAG00000000012"
"ENSBTAG00000000013" ...
..$ : chr [1:2] "PPEE" "PPDE"](https://image.slidesharecdn.com/rsemdepackages-180111065110/85/RSEM-and-DE-packages-38-320.jpg)
![Computational Biology and Genomics Facility, Indian Veterinary Research Institute
> DEfound=rownames(PP)[which(PP[,"PPDE"]>=.95)]
> str(DEfound)
chr [1:6528] "ENSBTAG00000000012" "ENSBTAG00000000013"
"ENSBTAG00000000015" "ENSBTAG00000000019"
"ENSBTAG00000000021" "ENSBTAG00000000025"
"ENSBTAG00000000026" "ENSBTAG00000000032" ...
> write.table(DEfound,"DE.txt",sep = "t",quote = F,col.names=F)
> GeneFC=PostFC(EBOut)
> write.table(GeneFC,"FC.txt",sep = "t",quote = F,col.names=F)
Output
GeneID PostFC Real FC comparison](https://image.slidesharecdn.com/rsemdepackages-180111065110/85/RSEM-and-DE-packages-39-320.jpg)











































































