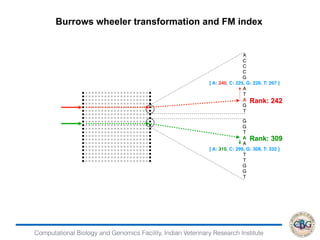
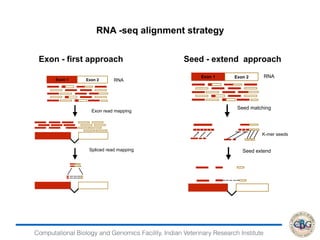
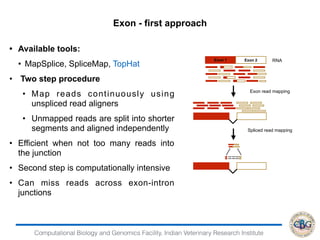
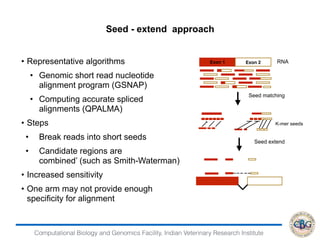
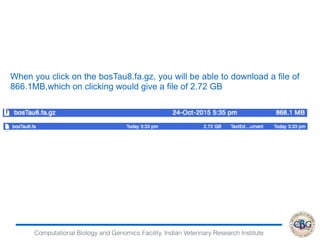
1. The document discusses various strategies for RNA-seq data analysis including fast sequence alignment strategies using hash tables and suffix trees.
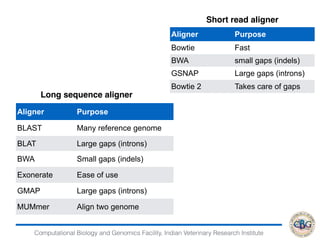
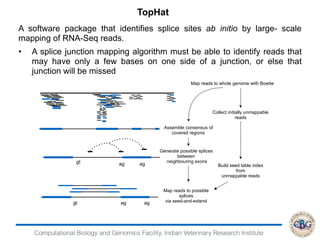
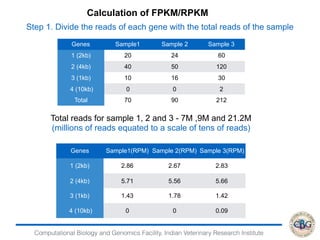
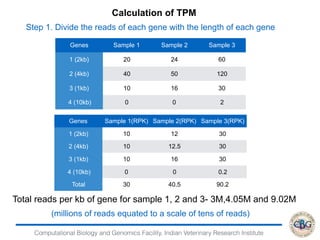
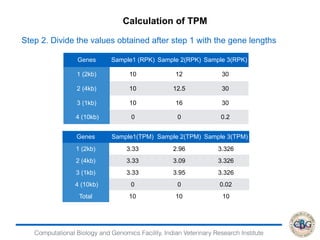
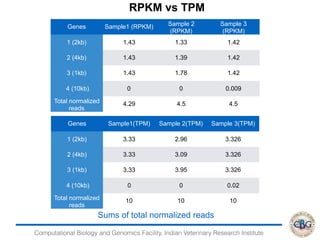
2. It also covers criteria for choosing aligners for different types of sequences, tools for mapping reads across splice junctions, and normalization methods like RPKM and TPM.
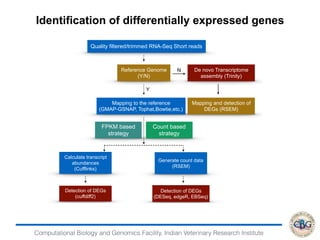
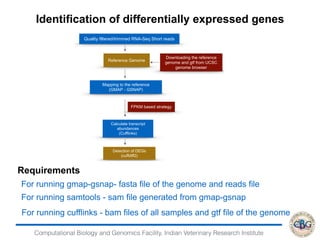
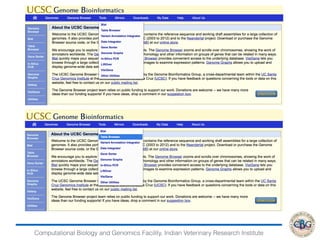
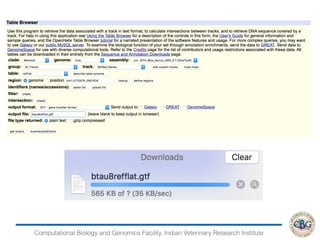
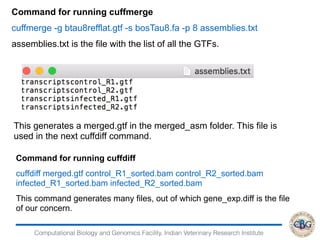
3. The key steps involved in the identification of differentially expressed genes from RNA-seq data using count-based and FPKM-based strategies are described. This includes mapping, quantifying transcripts, and using tools like Cufflinks, RSEM, DESeq and edgeR for detection of differentially expressed genes.