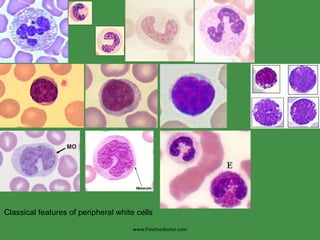
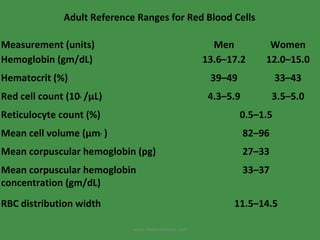
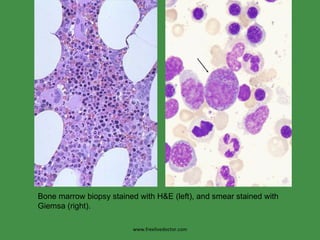
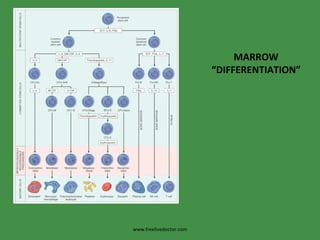
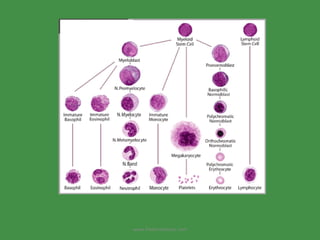
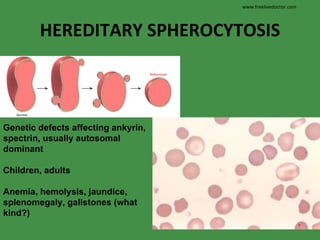
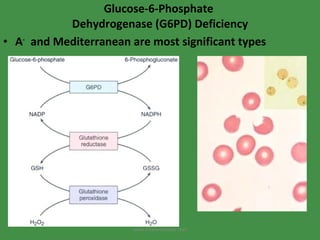
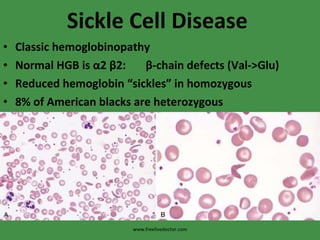
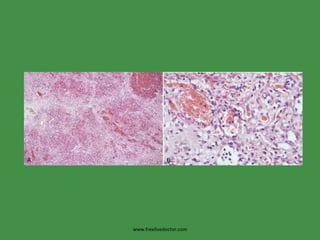
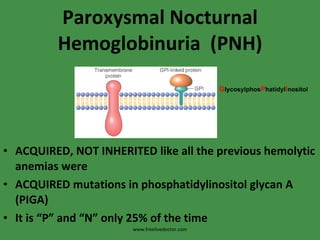
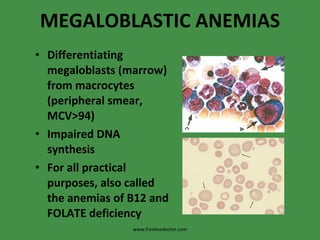
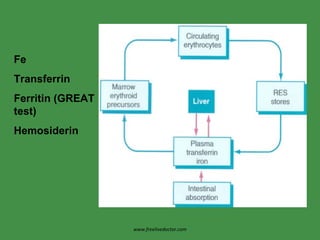
The document discusses red blood cells (RBCs) and bleeding disorders. It covers normal RBC anatomy, physiology and reference ranges. It then discusses various types of anemias including those from blood loss, decreased RBC production or increased destruction. Specific hemolytic anemias from hereditary disorders, enzymes deficiencies, antibodies and trauma are covered. Non-hemolytic anemias from B12/folate deficiencies, iron deficiency and bone marrow failure are also summarized. Polycythemia and bleeding disorders involving platelets, clotting factors and vessel walls are briefly mentioned.