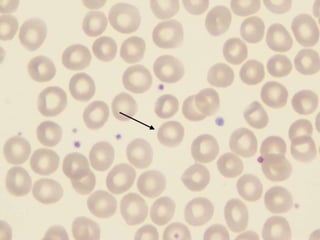
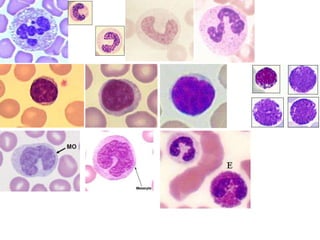
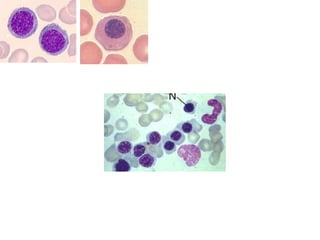
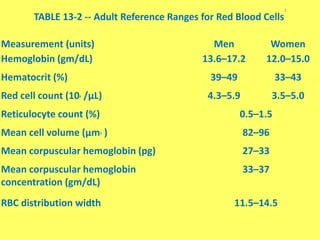
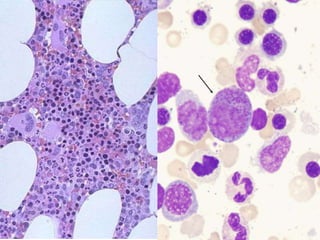
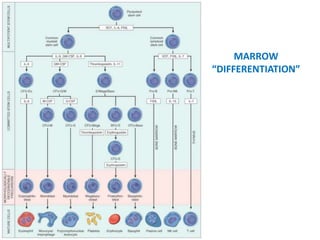
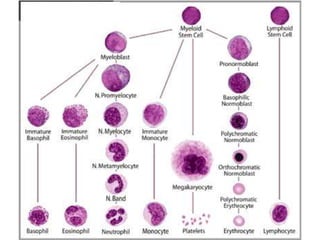
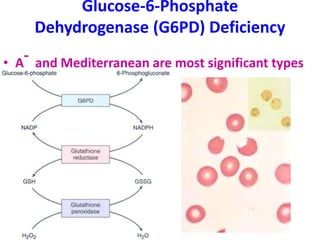
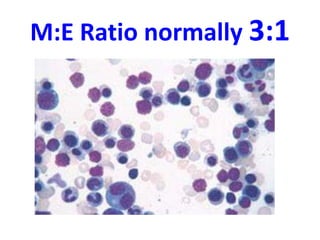
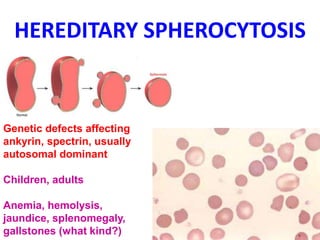
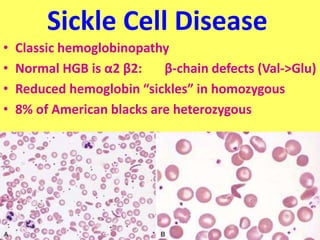
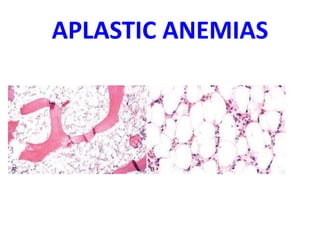
The document provides an extensive overview of red blood cells (RBCs) and bleeding disorders, detailing normal anatomy, types of anemias, and various conditions affecting RBC production and lifespan. It discusses hematologic features of different anemias, including blood loss, hemolytic diseases, and production impairments, alongside a variety of bleeding disorders such as thrombocytopenias and hemophilias. Additionally, it covers diagnostic tests, treatment options, and implications of conditions like DIC and thrombosis.