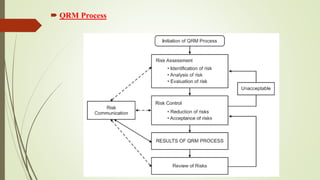
The document discusses Quality Risk Management (QRM) in the pharmaceutical industry, emphasizing the importance of assessing and controlling quality variations throughout a product's lifecycle. It details the systematic QRM process, including initiation, risk assessment, control, reviews, communication, and necessary information for technology transfer, active pharmaceutical ingredients, excipients, and finished products. Documentation and equipment considerations are also highlighted as essential for effective technology transfer and product quality assurance.


























































































![谷歌留痕技术 [ 𝙩𝙤𝙥 𝟮𝟯𝟯. 𝙘 𝙤𝙢 ]](https://cdn.slidesharecdn.com/ss_thumbnails/top233-260130174328-3833018c-thumbnail.jpg?width=640&height=640&fit=bounds)












