Downloaded 10 times


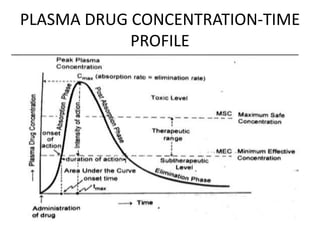




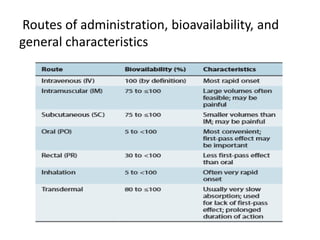



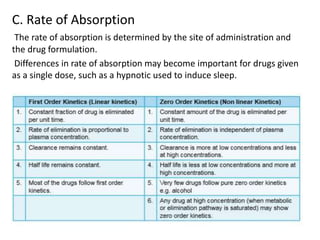
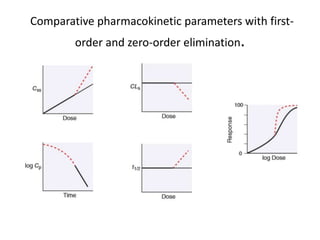






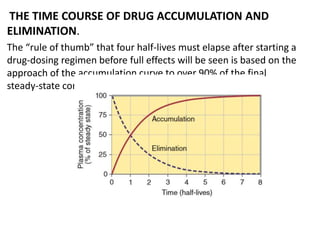
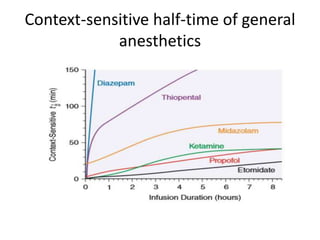


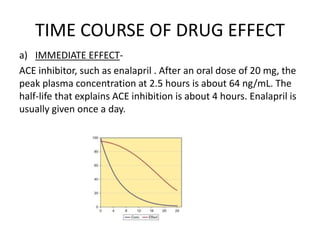



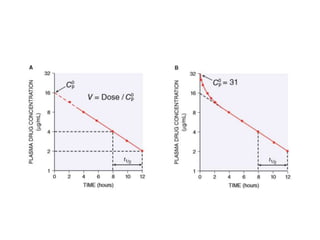



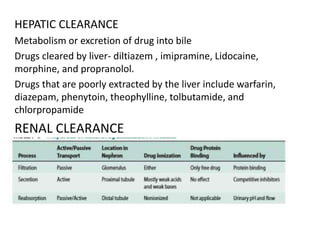







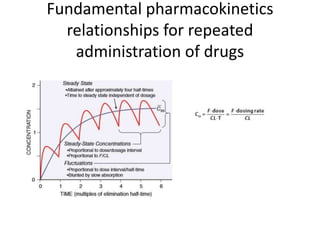







This document discusses pharmacokinetic parameters essential for understanding drug absorption and efficacy, including peak plasma concentration (Cmax), time of peak concentration (Tmax), and area under the curve (AUC). It highlights the implications of drug bioavailability, half-life, clearance, and the effects of first-pass metabolism, along with practical examples of various medications. Special attention is given to the pharmacokinetic characteristics that influence dosing regimens and potential drug interactions.



































![Advanced practice preparation pharmacodynamics[1]](https://cdn.slidesharecdn.com/ss_thumbnails/advancedpracticepreparationpharmacodynamics1-120402100026-phpapp02-thumbnail.jpg?width=640&height=640&fit=bounds)






![Clinical Pharmacokinetics-I [half life, order of kinetics, steady state]](https://cdn.slidesharecdn.com/ss_thumbnails/clinicalpk-ihalflifeorderofkineticssteadystate-140217020044-phpapp02-thumbnail.jpg?width=640&height=640&fit=bounds)





![PERI-PROSTHETIC FRACTURE NAIL-PLATE CONSTRUCT [NPC].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/drarunkumardrmohamedashrafperiprostheticfrasturenail-plateconstructnpc-260209164459-7e9d15a1-thumbnail.jpg?width=640&height=640&fit=bounds)
















![CTEV [ clubfoot] DR ARUN LAL ,DR MOHAMED ASHRAF travancore medical college k...](https://cdn.slidesharecdn.com/ss_thumbnails/ctevclubfootdrarunlaldrmohamedashraftravancoremedicalcollegekollamkeralaindia-260208063247-18fc466c-thumbnail.jpg?width=640&height=640&fit=bounds)

