Download to read offline



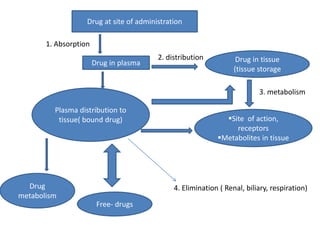




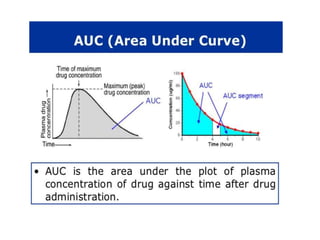
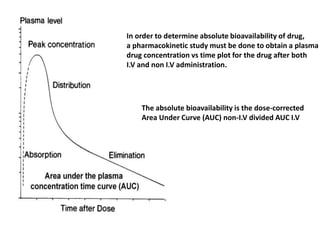
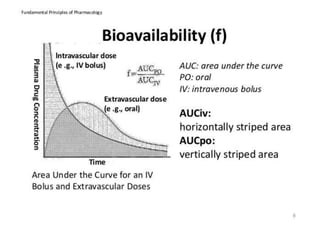









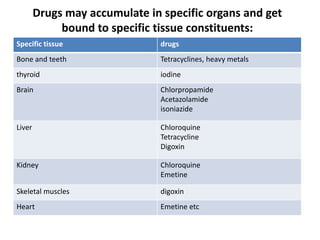












This document discusses various pharmacokinetic concepts including absorption, distribution, metabolism, and excretion of drugs. It describes how drugs are absorbed through various routes and distributed throughout the body. Factors that influence bioavailability such as drug properties, formulation, and first-pass metabolism are examined. The document also explores drug metabolism through phase I and II reactions in the liver and how metabolites are excreted primarily through the kidneys or feces. Teratogenic drugs and drugs contraindicated during pregnancy or breastfeeding are highlighted.








![Clinical Pharmacokinetics-I [half life, order of kinetics, steady state]](https://cdn.slidesharecdn.com/ss_thumbnails/clinicalpk-ihalflifeorderofkineticssteadystate-140217020044-phpapp02-thumbnail.jpg?width=640&height=640&fit=bounds)


































































![PERI-PROSTHETIC FRACTURE NAIL-PLATE CONSTRUCT [NPC].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/drarunkumardrmohamedashrafperiprostheticfrasturenail-plateconstructnpc-260209164459-7e9d15a1-thumbnail.jpg?width=640&height=640&fit=bounds)











