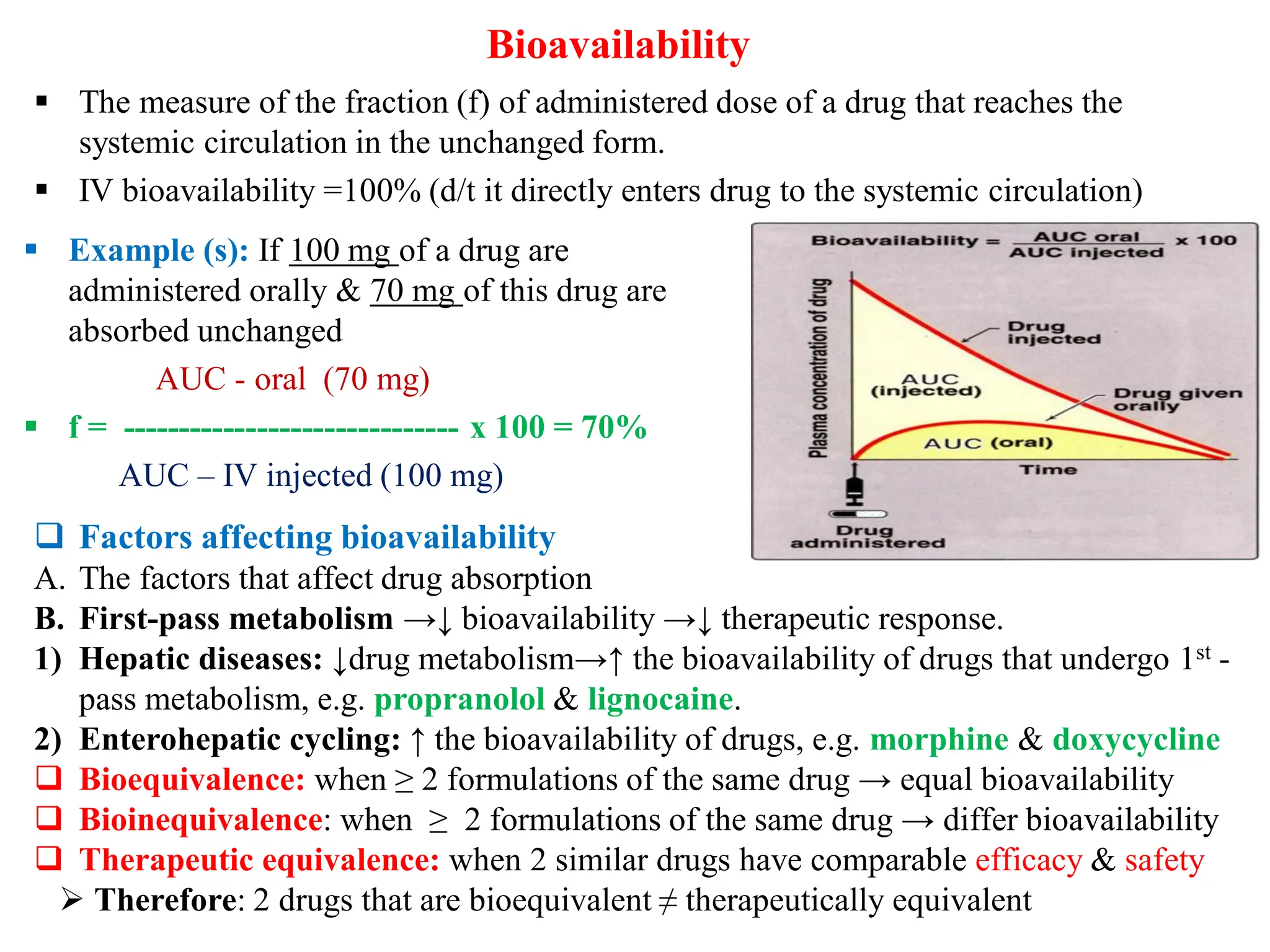
The document discusses pharmacokinetics and pharmacodynamics, covering key concepts such as absorption, distribution, metabolism, and excretion (ADME) of drugs. It outlines the factors influencing drug absorption and distribution, including physicochemical properties and patient-related factors, as well as the role of plasma protein binding in drug action. The document highlights the importance of first-pass metabolism and bioavailability in understanding drug efficacy and therapeutic responses.









![Factors influencing drug absorption
A. Physicochemical properties of the drug
Liquid form of the drug is better absorbed than solid form
1) Physical state
Shorter the time, better is the absorption
2) Disintegration &
dissolution times
Lipid-soluble & unionized form of the drug is better absorbed
than the water-soluble & ionized form.
3) Degree of
ionization &
lipid solubility
Inorganic small molecules are better absorbed than organic larger
drugs
E.g. microfine aspirin (well absorbed) , anthelmintics (larger
particle size) → poorly absorbed from GI tract → better effect on
gut helminthes]
4) Chemical nature
& MW
Inert substances (lactose, starch, Ca sulphate, gum) are added to
formulations as binding agents→ affect the absorption of drugs
(e.g. Ca+2 → ↓ absorption of tetracyclines)
5) Formulations
(dosage forms)
Ferrous form (Fe2+ ) is more absorbed than ferric form (Fe3+)
6) Valency
<
< <](https://image.slidesharecdn.com/lp-2pharmacokineticsfinal-241027205524-519ea62c/75/L-P-2-Pharmacokinetics-final_-pdf-10-2048.jpg)










![2. Placental barrier:
Lipid cellular barrier between mother & fetus
Lipid soluble, non-ionized drugs can pass easily, (e.g., methimazole]
Non-lipid soluble drugs (e.g., propylthiouracil ), quaternary ammonium
compounds, e.g. d- tubocurarine (d-TC) & substances with high MW like
insulin cannot cross the placental barrier
Has some influx transporters, & placental efflux P-gp (limit foetal exposure to
maternally administered drugs)
More permeable than BBB → almost any drug taken by the mother can affect
the foetus/or the newborn.
o Drug taken in the 1st - trimester as phenytoin & tetracyclines→ teratogenicity
/or malformations
o Drugs taken during labor as morphine & barbiturates → neonatal asphyxia](https://image.slidesharecdn.com/lp-2pharmacokineticsfinal-241027205524-519ea62c/75/L-P-2-Pharmacokinetics-final_-pdf-22-2048.jpg)

































![L P 1 Antihypertensive agents- -final-2022 [وضع التوافق]-1.en.ar.pdf](https://cdn.slidesharecdn.com/ss_thumbnails/lp1antihypertensiveagents-final-2022-1-241009070554-2f460b7b-thumbnail.jpg?width=640&height=640&fit=bounds)





























