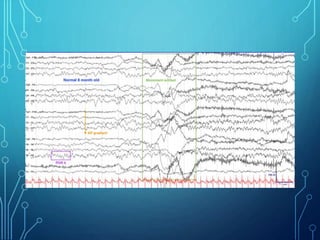
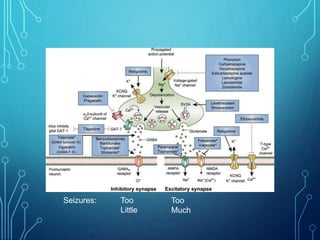
- Infantile spasms, also known as West syndrome, is a specific type of epilepsy seen in infants characterized by infantile spasms, hypsarrhythmia on EEG, and developmental regression or delay.
- It represents 2% of epilepsies and typically presents between 4-6 months of age. The condition was first described in 1841 and involves sudden flexion or extension of the trunk and limbs.
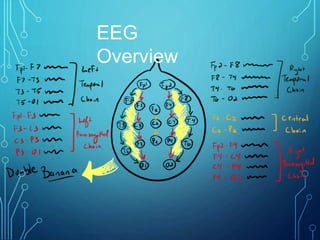
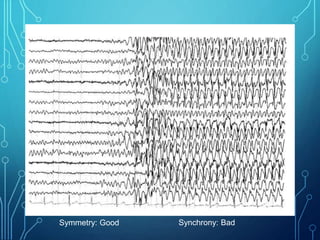
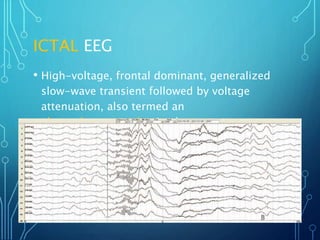
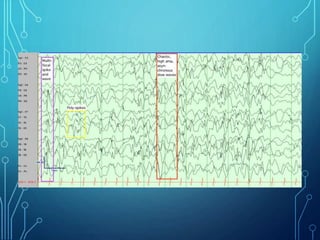
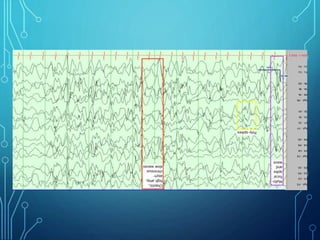
- Evaluation involves neurological exam, imaging (often MRI), metabolic testing, and characteristic EEG findings of hypsarrhythmia. Treatment aims to stop spasms and normalize EEG typically within 2-4 weeks using ACTH/steroids as first line. Prognosis depends on





































![TREATMENT
• First-line treatments (ie, ACTH, prednisone,
vigabatrin, pyridoxine [vitamin B-6])
• USA natural, porcine ACTH (ACTHAR) -- $$$$$$
• Europe tetracosactide, synthetic -- $
• Second-line treatments (ie, benzodiazepines,
valproic acid, lamotrigine, topiramate,
zonisamide)
• Combo?](https://image.slidesharecdn.com/infantilespasms-creed-231024212150-ef34837a/85/Infantile-Spasms-ppt-44-320.jpg)









































































![CASE_PRESENTATION_ON_subdural_hematoma(SDH)[1 FINAL PPT]-1.pptx](https://cdn.slidesharecdn.com/ss_thumbnails/casepresentationonsubduralhematomasdh1finalppt-1-260129172522-d405d375-thumbnail.jpg?width=640&height=640&fit=bounds)