Downloaded 11 times



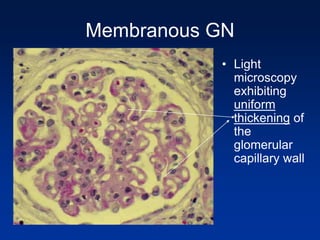
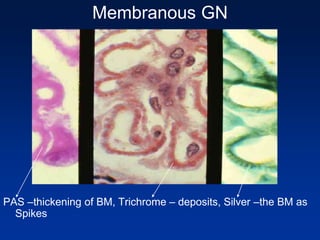

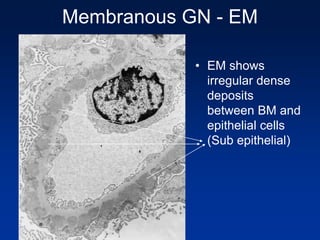



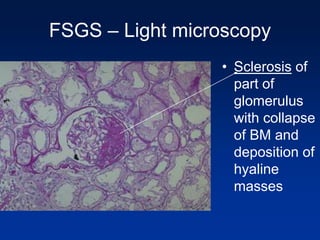
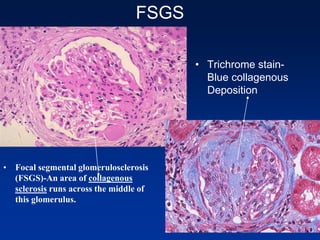
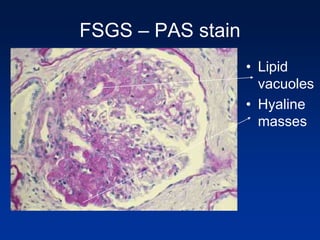

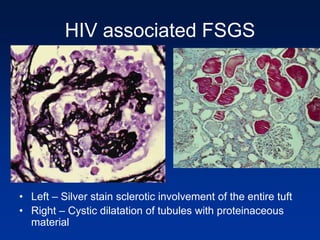





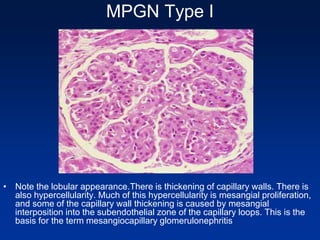
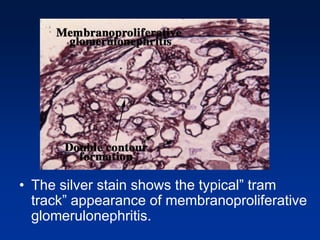
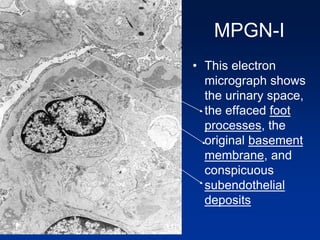
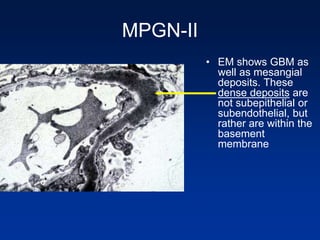



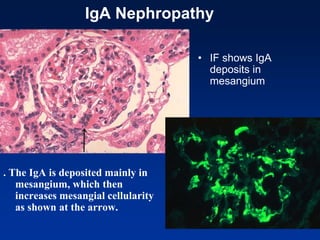



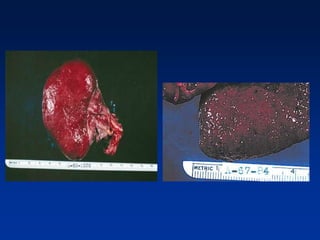
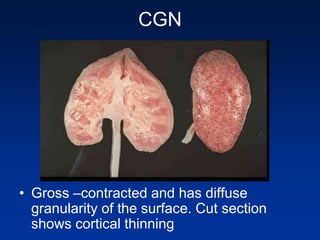
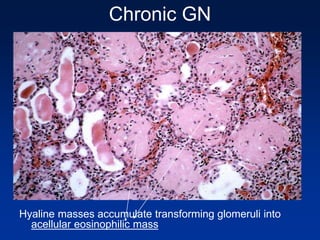


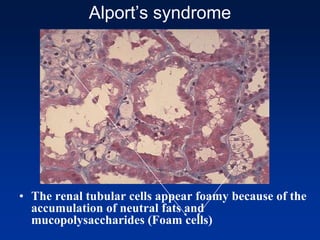






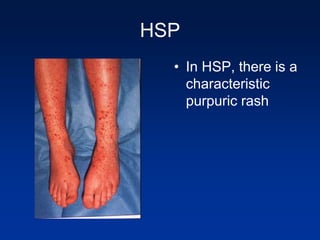





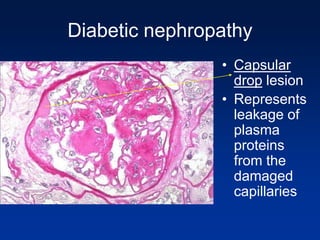

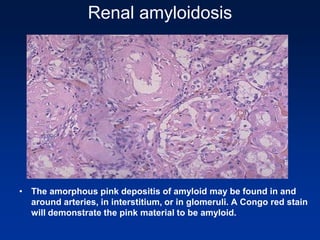
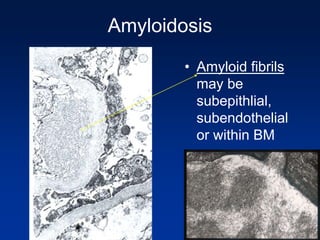


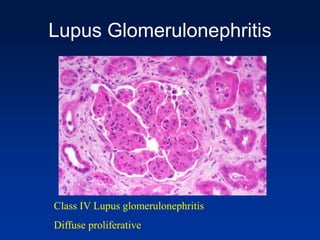
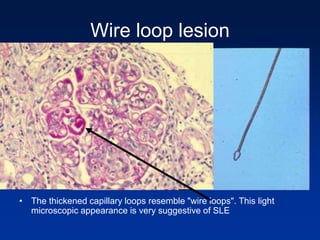
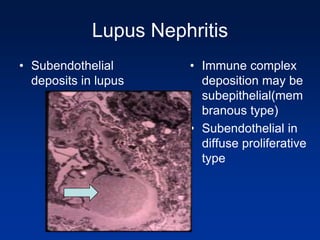



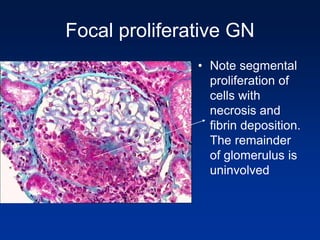

The document provides an overview of various forms of glomerulonephritis (GN), highlighting their causes, pathogenesis, histological features, clinical courses, and associated systemic diseases. It covers conditions such as membranous GN, focal segmental glomerulosclerosis (FSGS), membranoproliferative GN, IgA nephropathy, and others, discussing the implications of each type on renal health and prognosis. The document emphasizes the significance of underlying causes, especially infections and autoimmune diseases, in the development of these renal disorders.






















































































