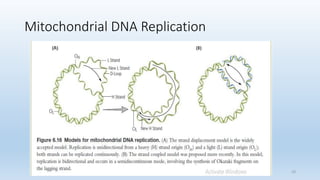
Mitochondria are semi-autonomous organelles found in eukaryotic cells that are thought to have originated from endosymbiotic bacteria. They contain their own circular genome and are the site of aerobic respiration and ATP production through oxidative phosphorylation. Mitochondrial DNA is maternally inherited and mutations can cause a variety of mitochondrial diseases due to defects in energy production.










































![extra-chromosomal-inheritance[1].pptx.pdfpdf](https://cdn.slidesharecdn.com/ss_thumbnails/extra-chromosomal-inheritance1-240529182826-8153c739-thumbnail.jpg?width=640&height=640&fit=bounds)

































































