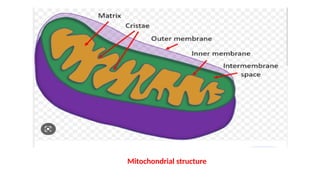
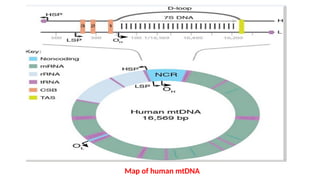
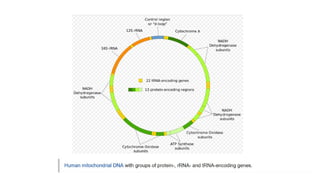
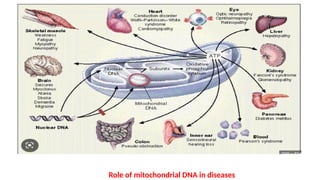
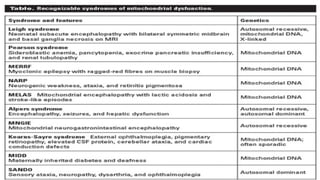
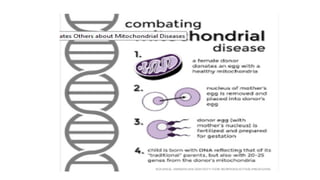
Mitochondria are essential organelles in eukaryotic cells responsible for energy production and various cellular processes, containing their own circular DNA (mtDNA) that encodes 13 proteins and is maternally inherited. Mitochondrial diseases, which are genetically heterogeneous, arise from mutations in mtDNA or nuclear DNA, often causing significant clinical symptoms across multiple organ systems. Current treatment strategies are largely symptomatic, with emerging therapies like gene editing and mitochondrial donation showing promise but facing challenges in effectiveness and ethical considerations.