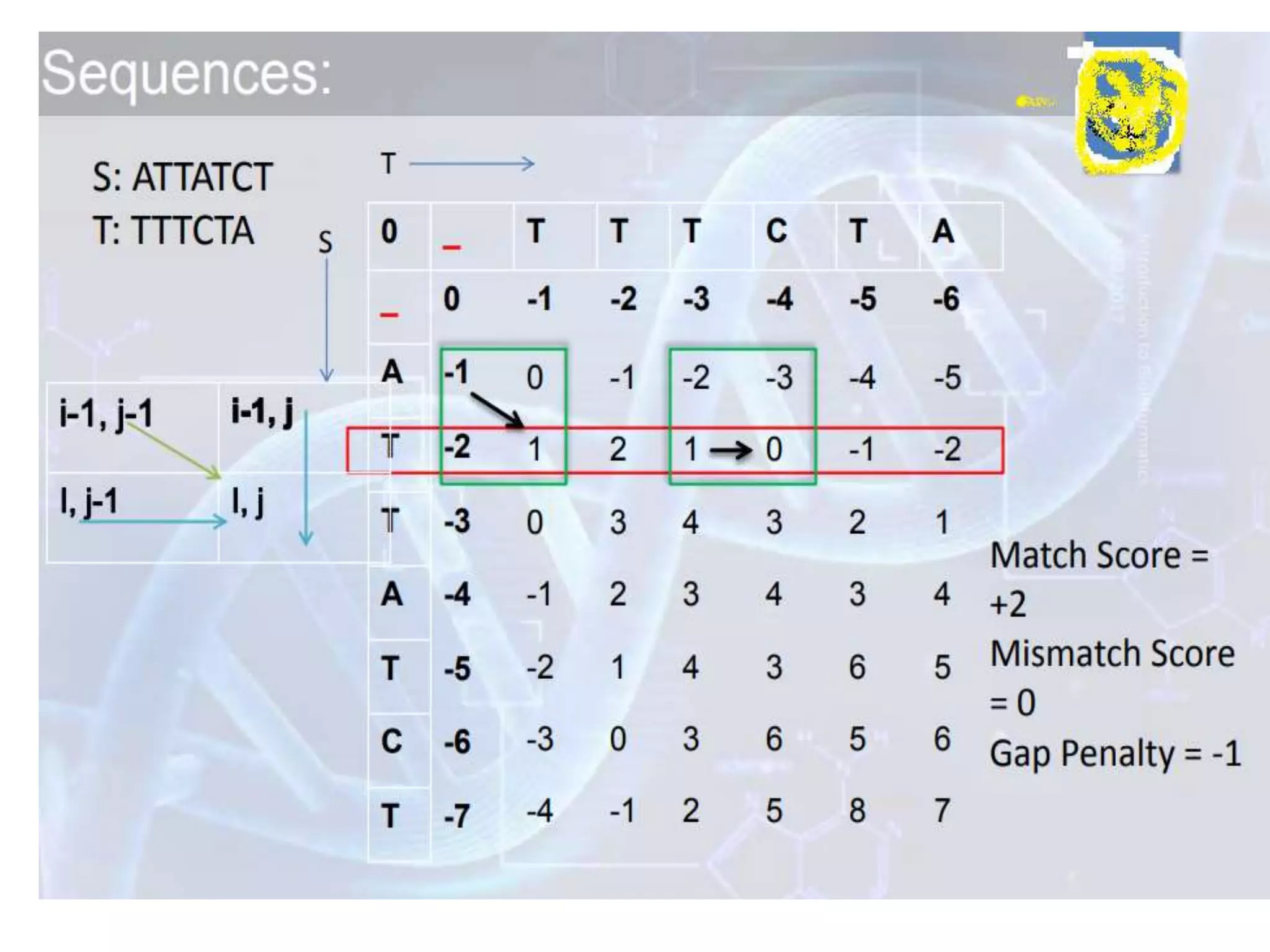
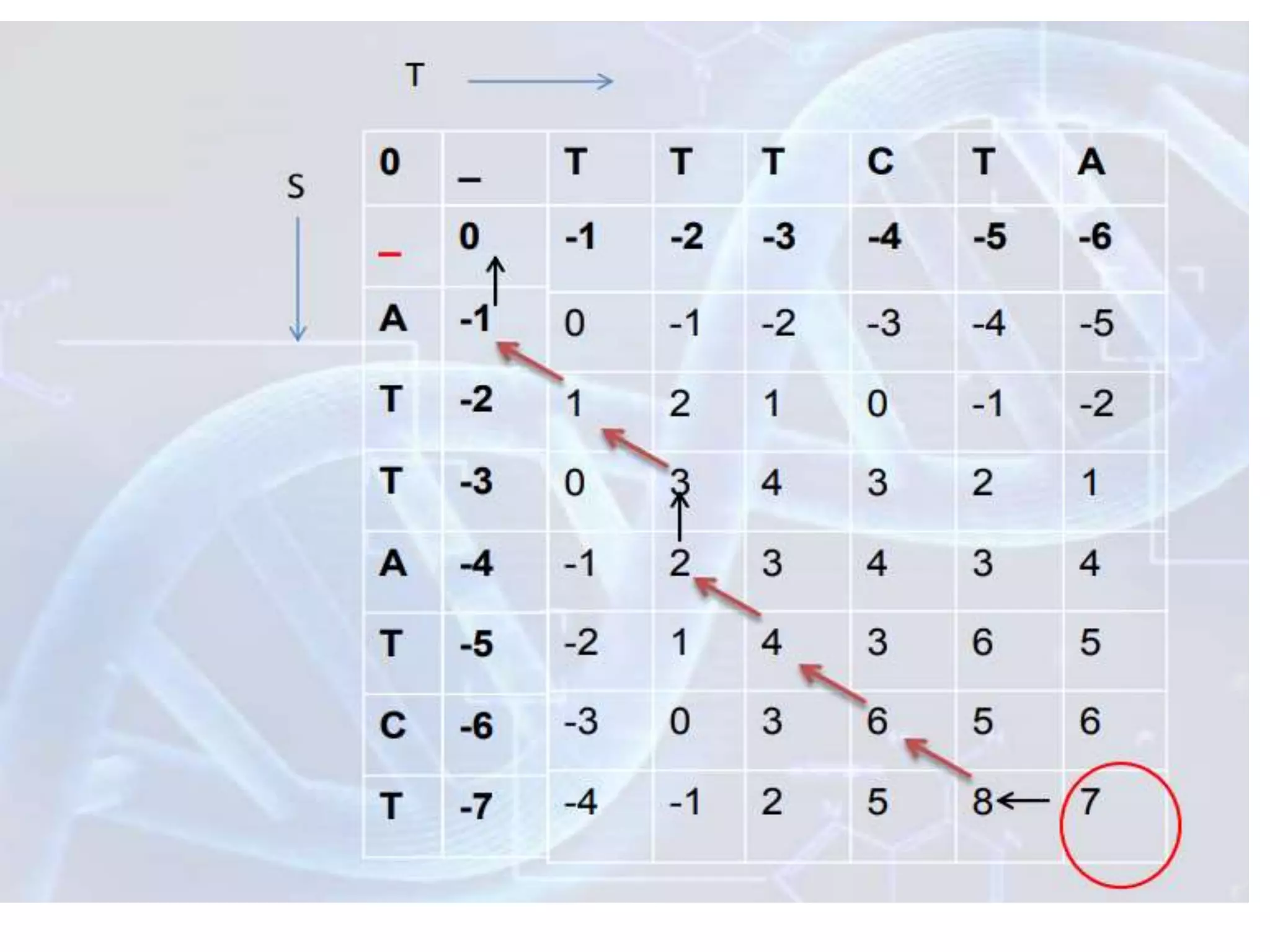
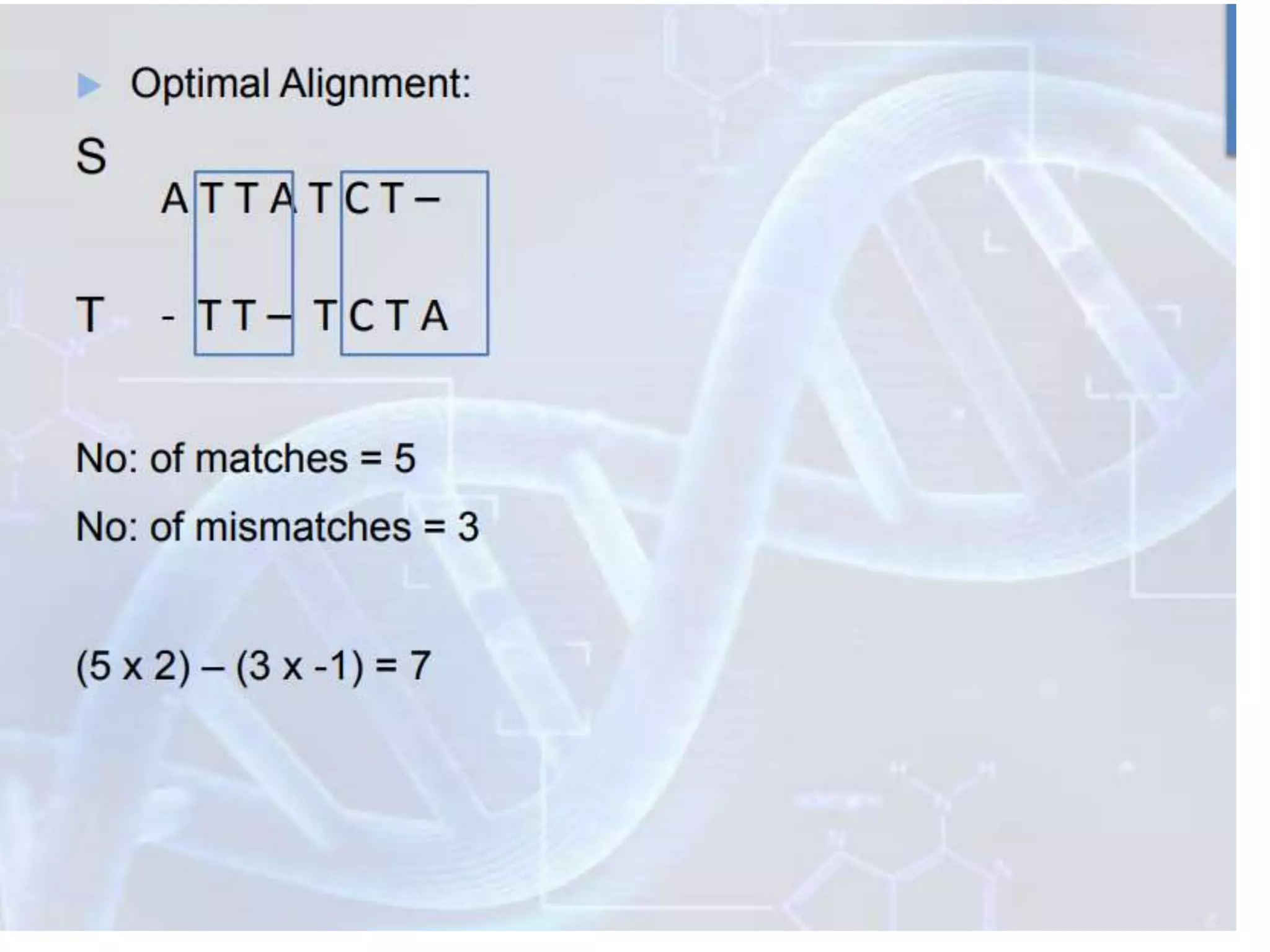
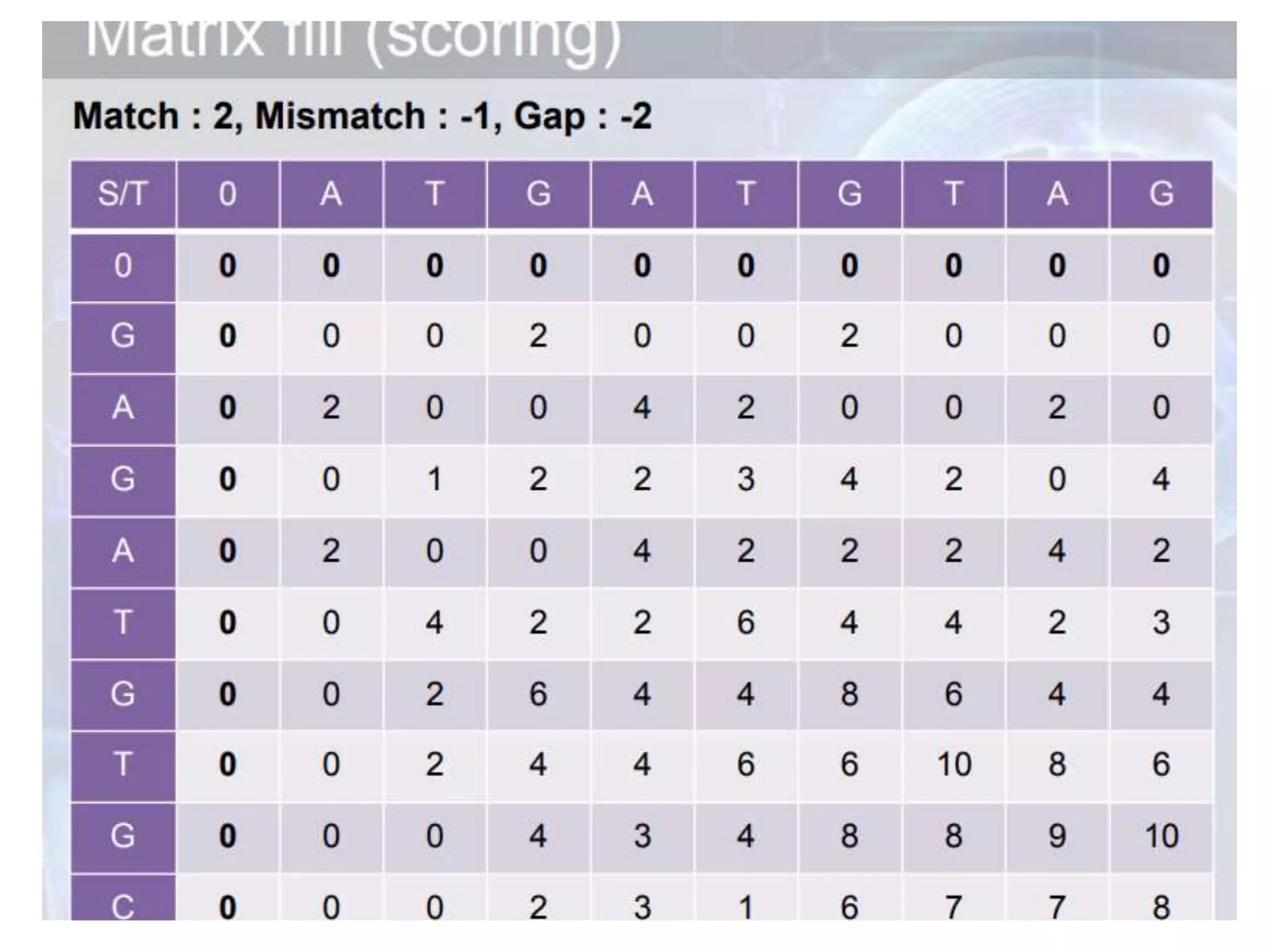
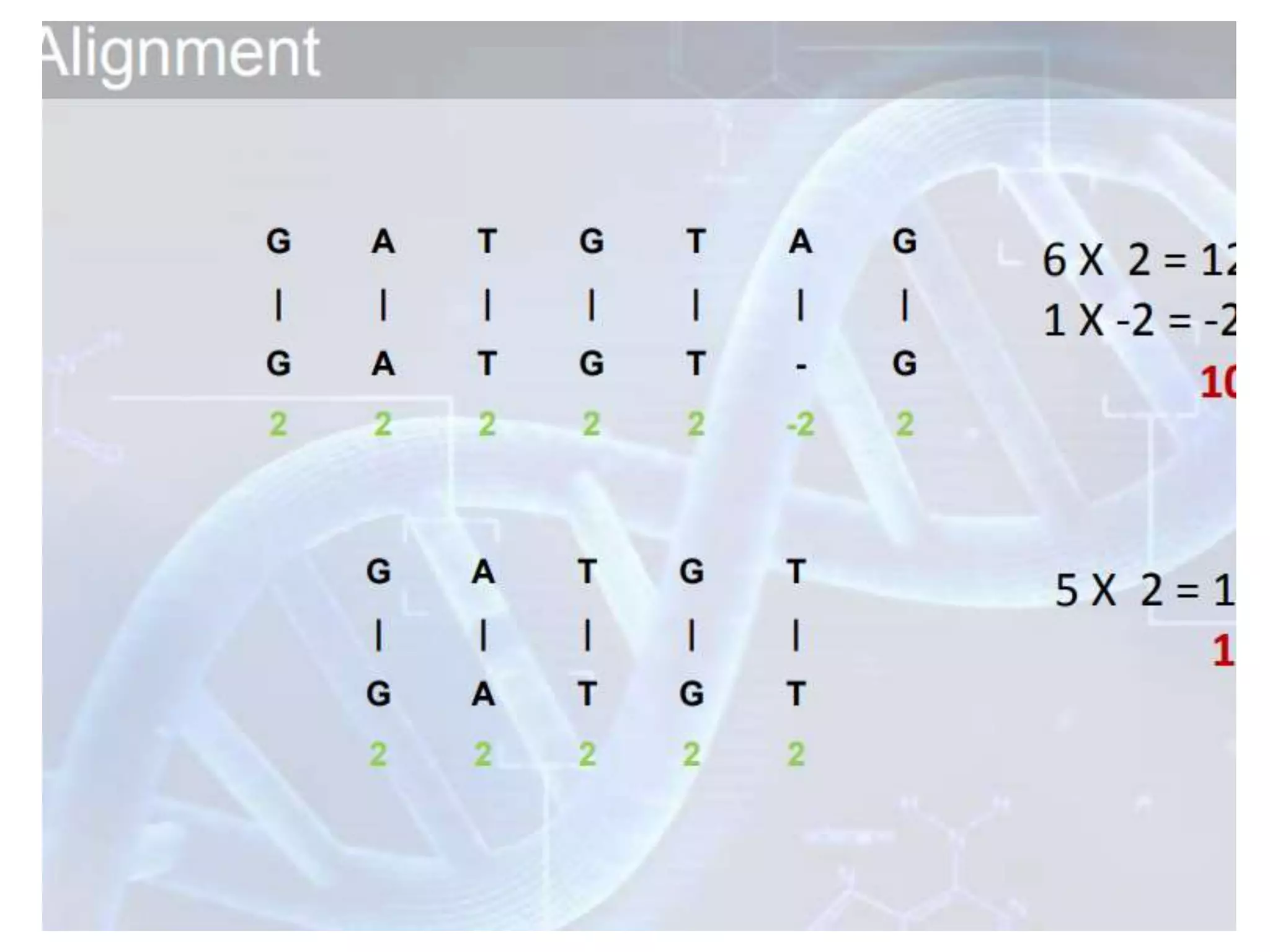
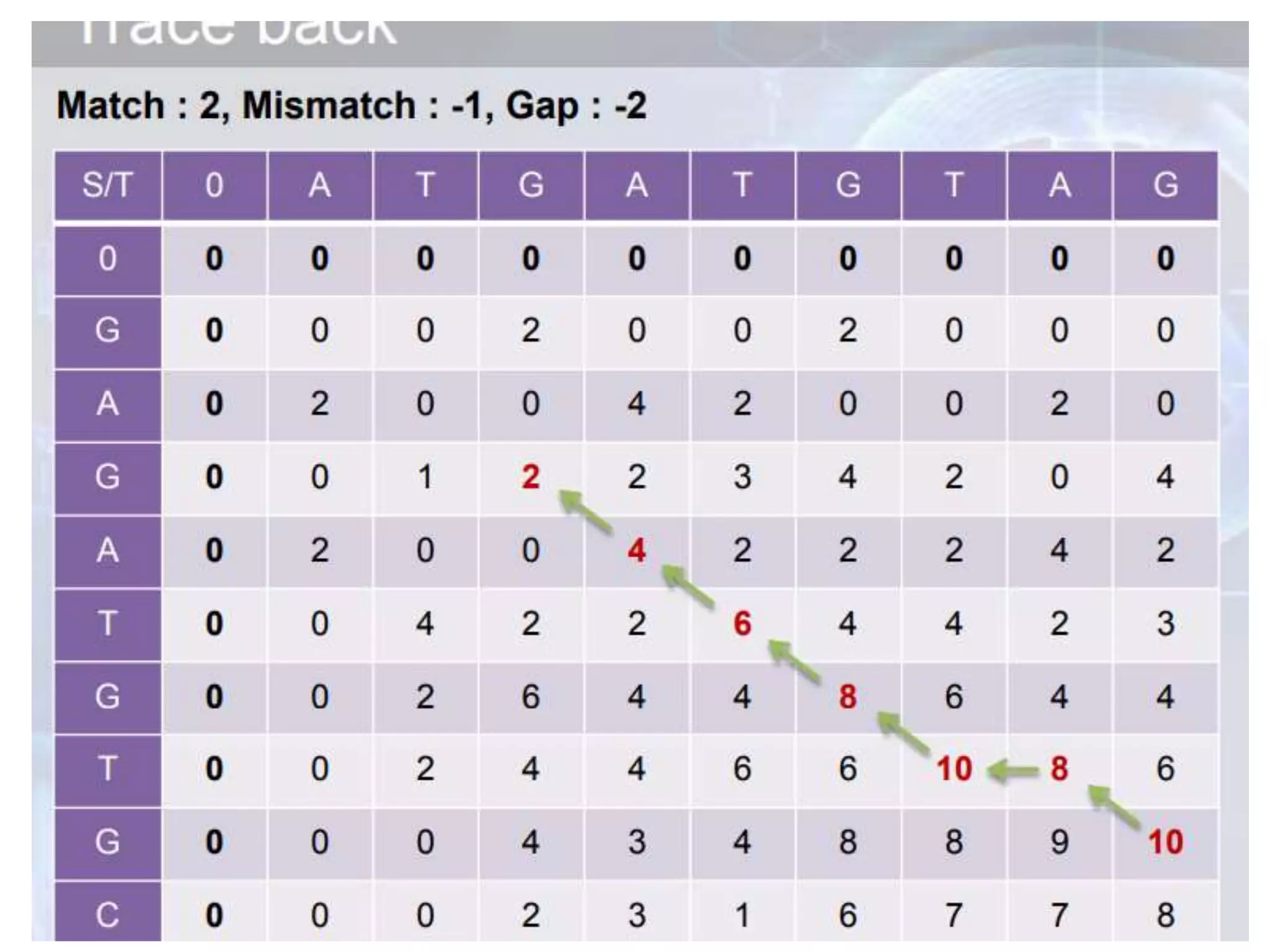
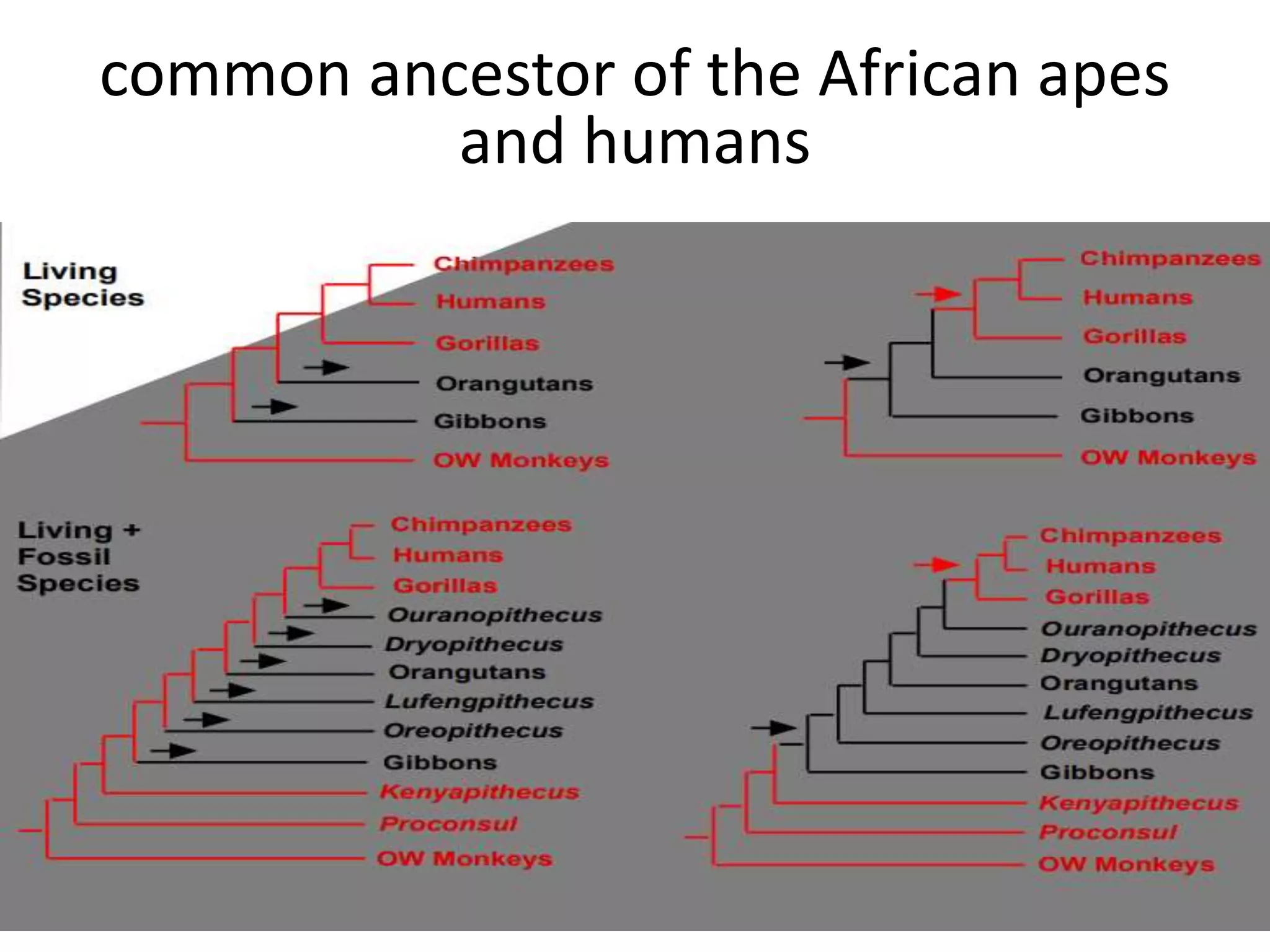
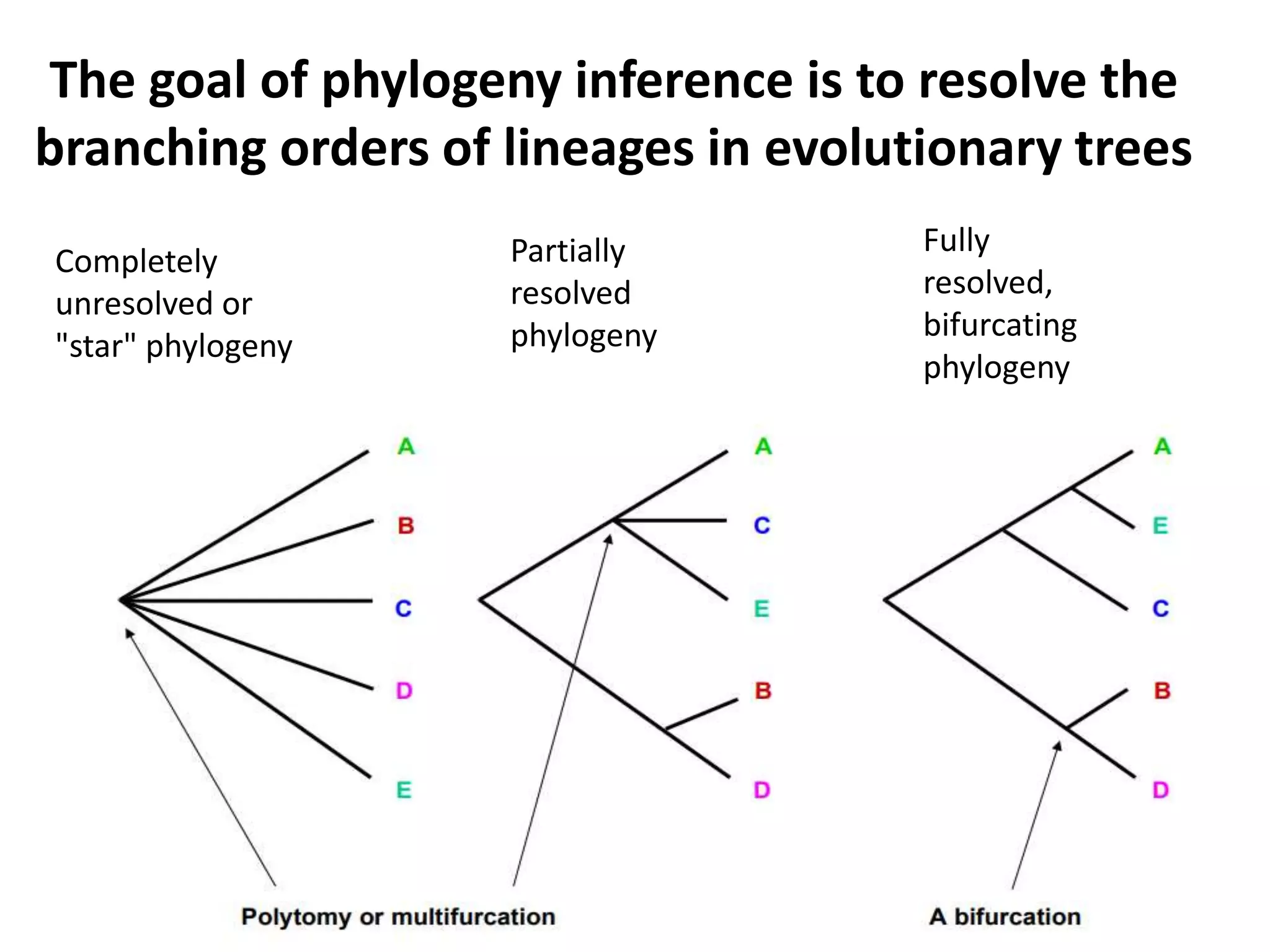
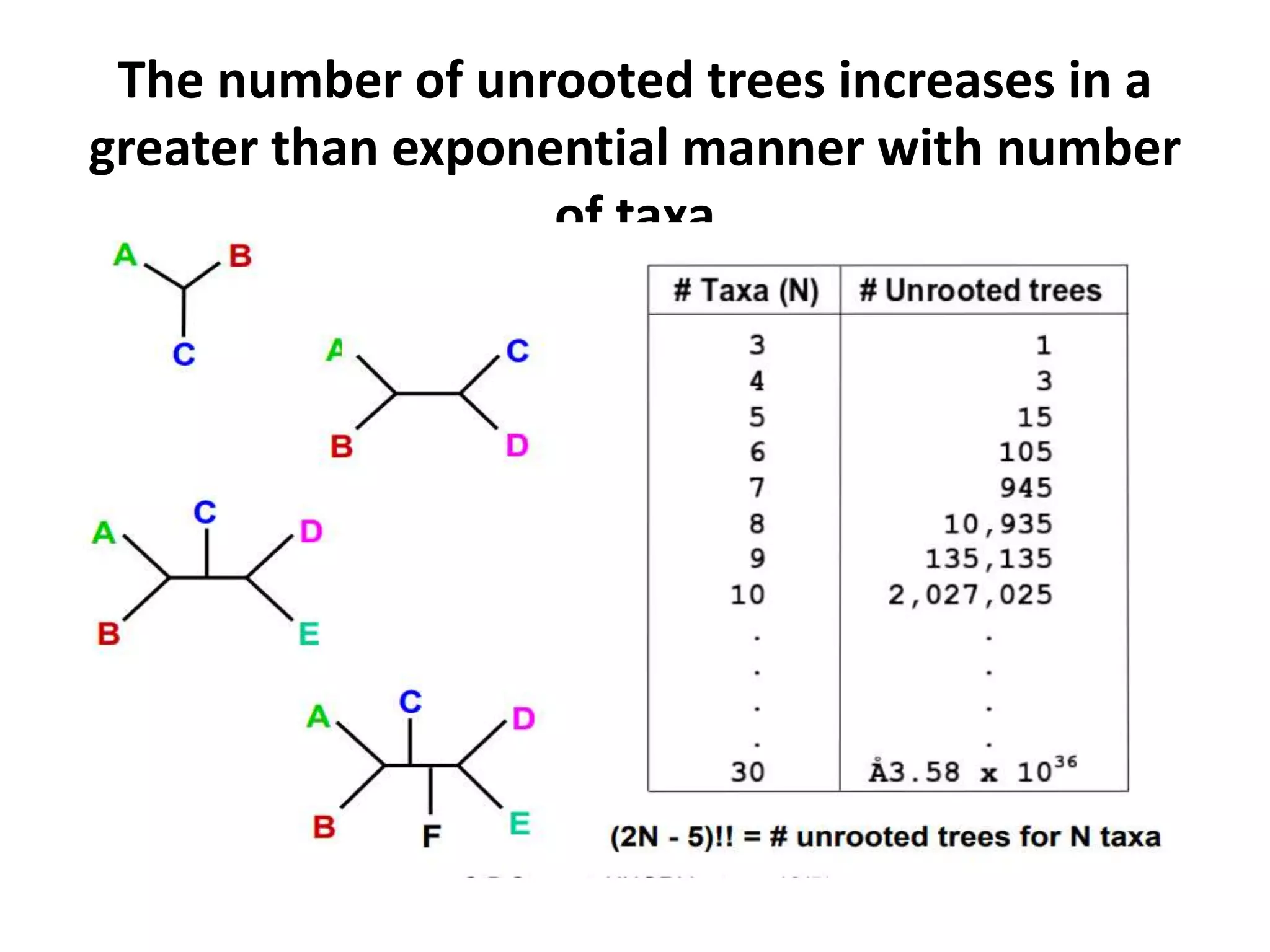
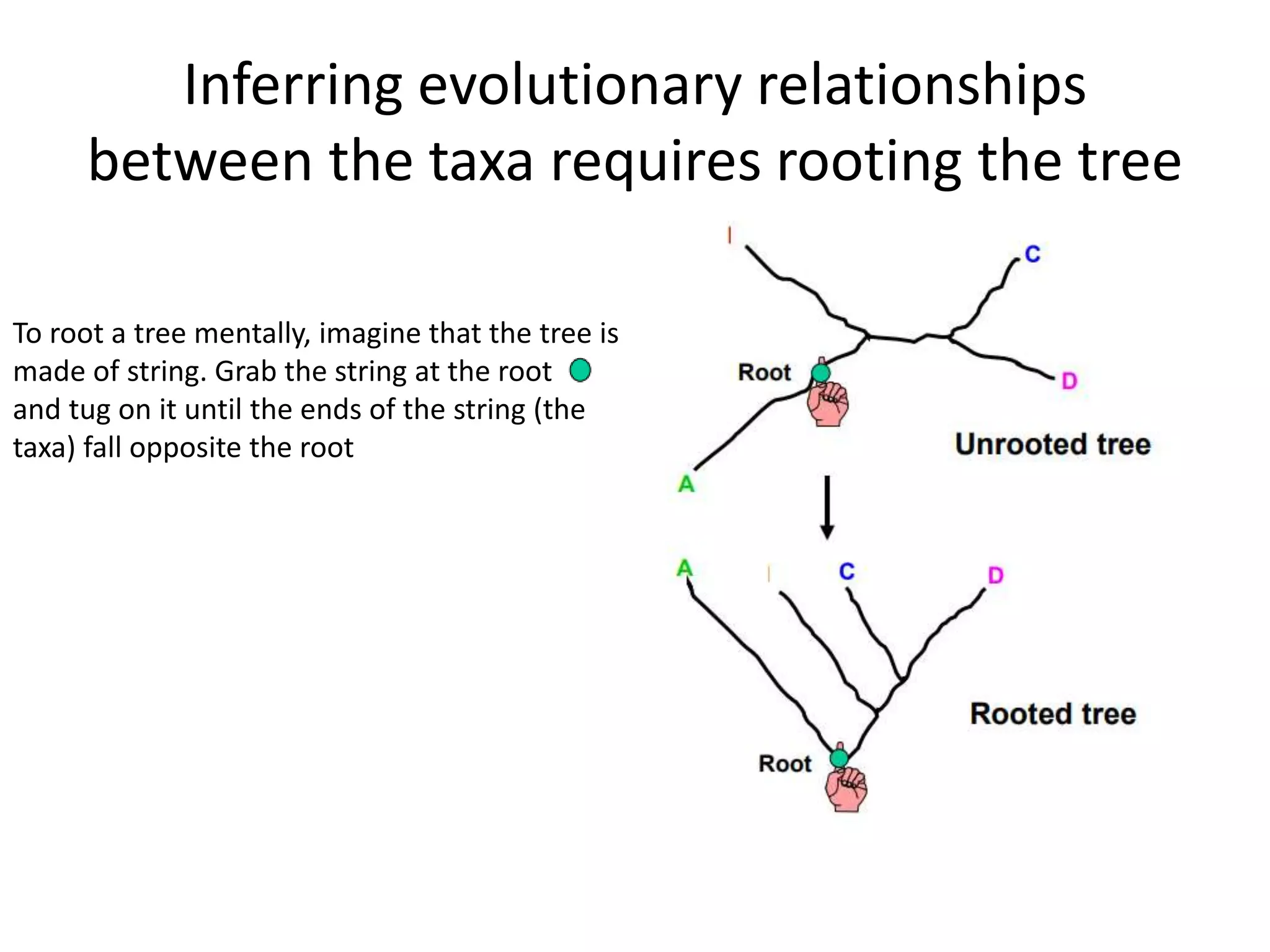
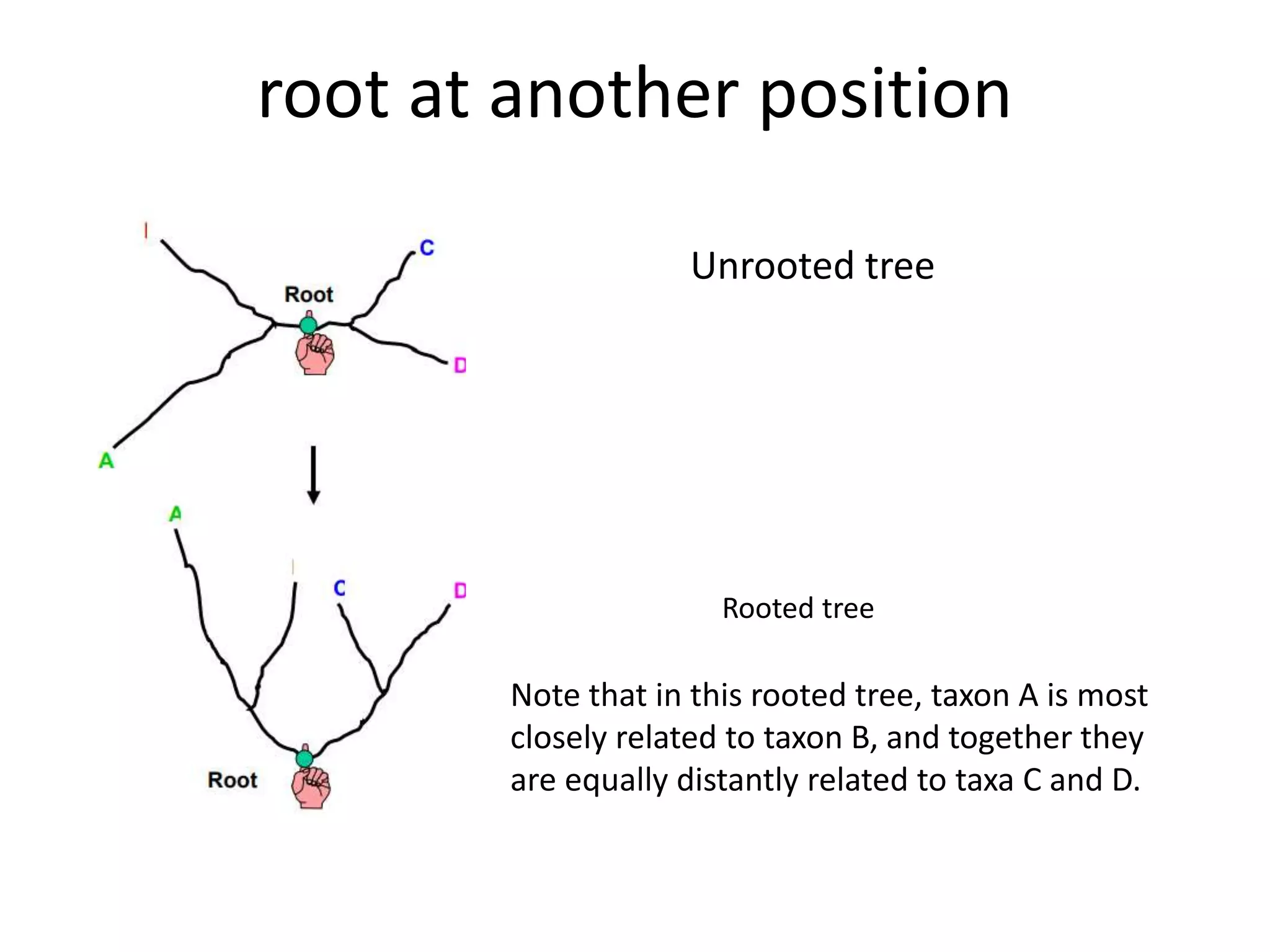
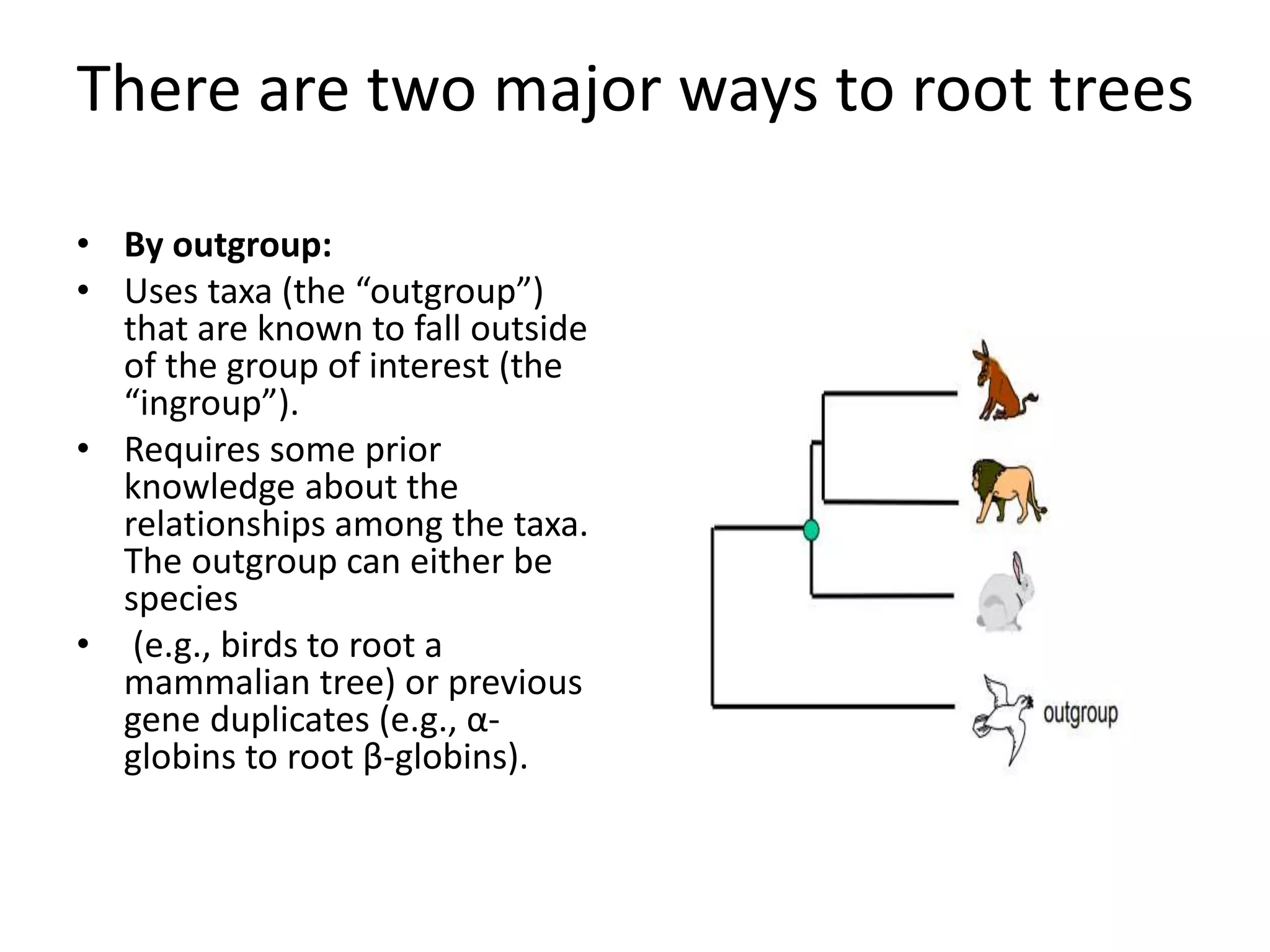
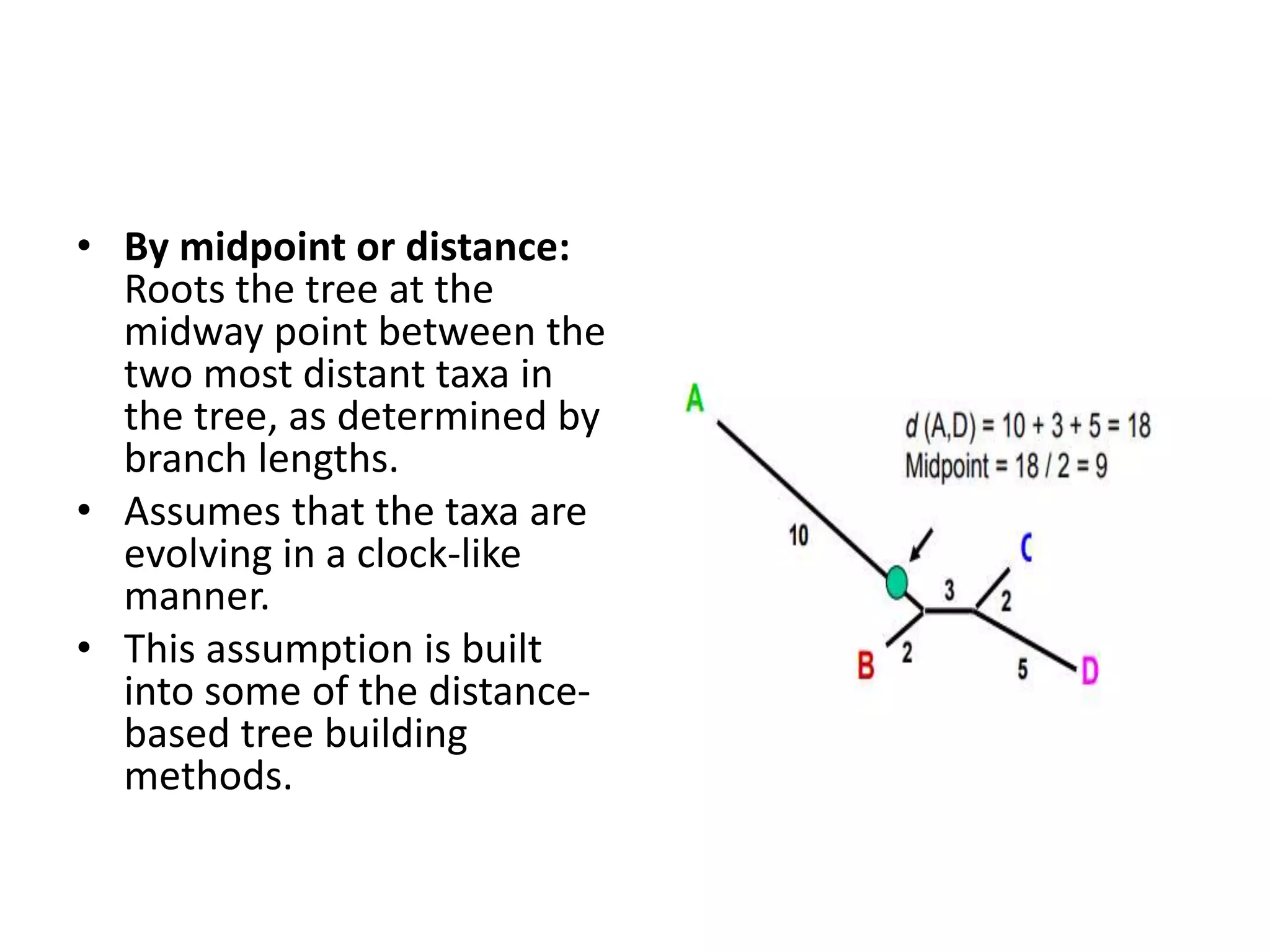
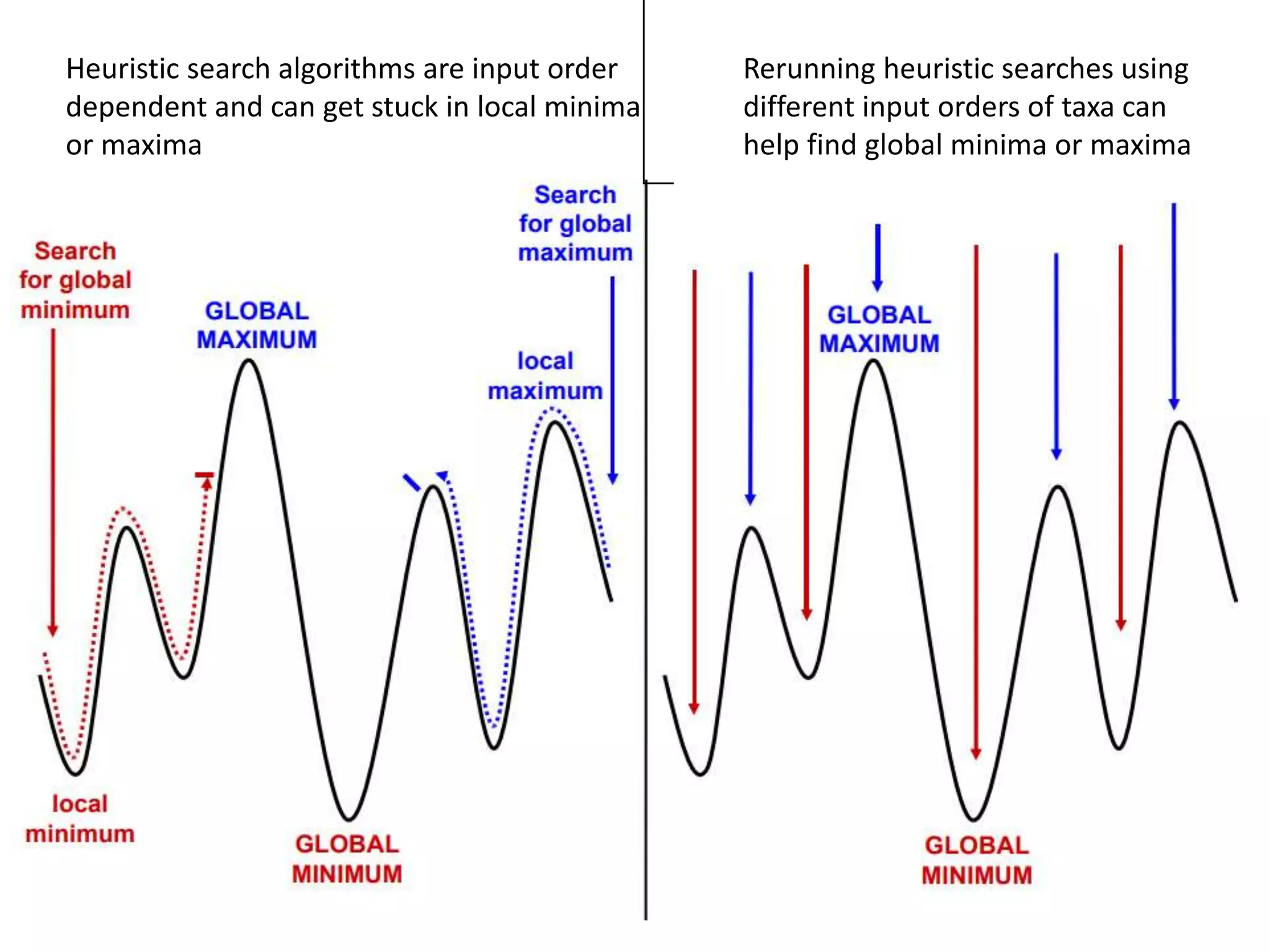
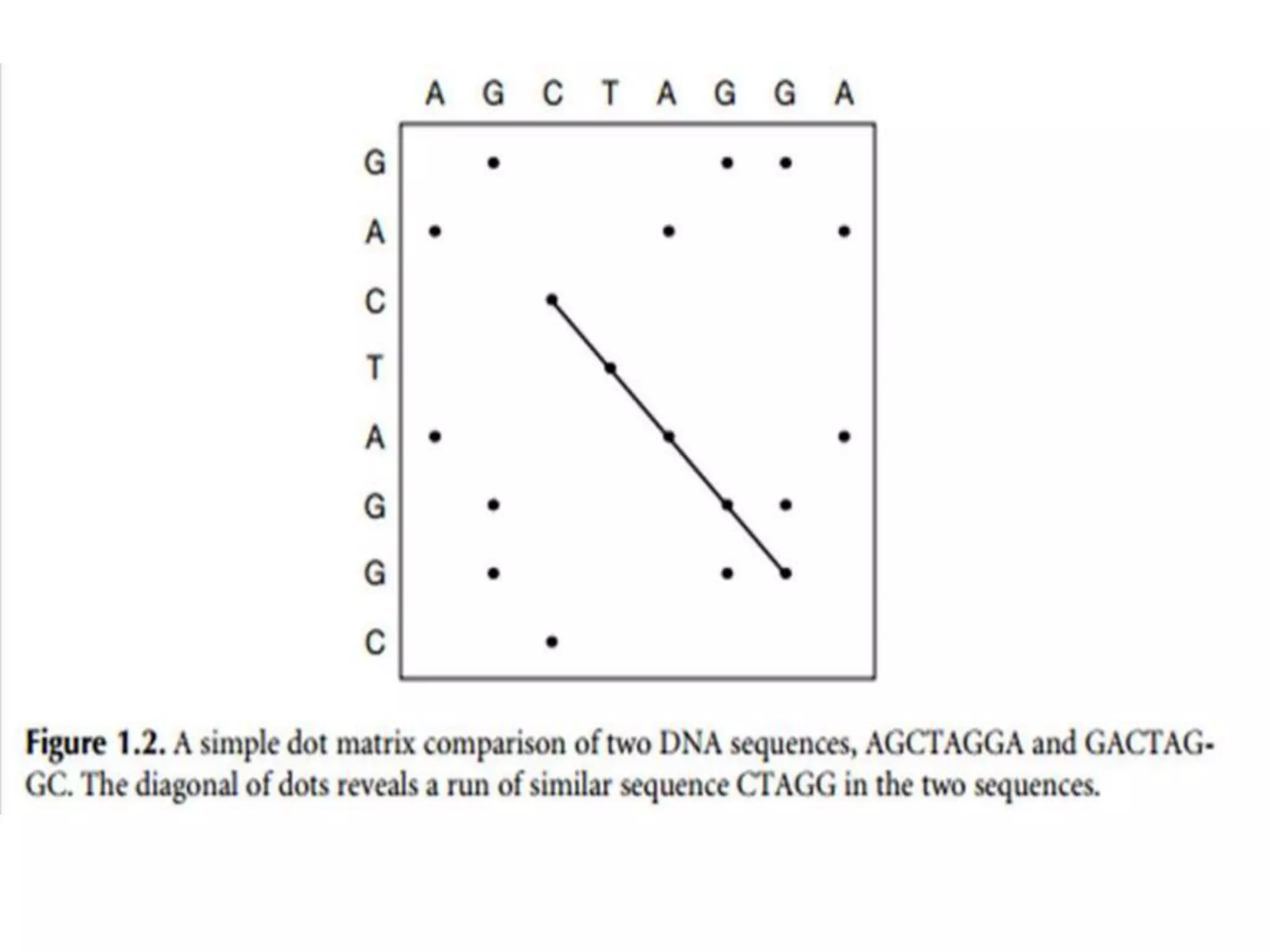
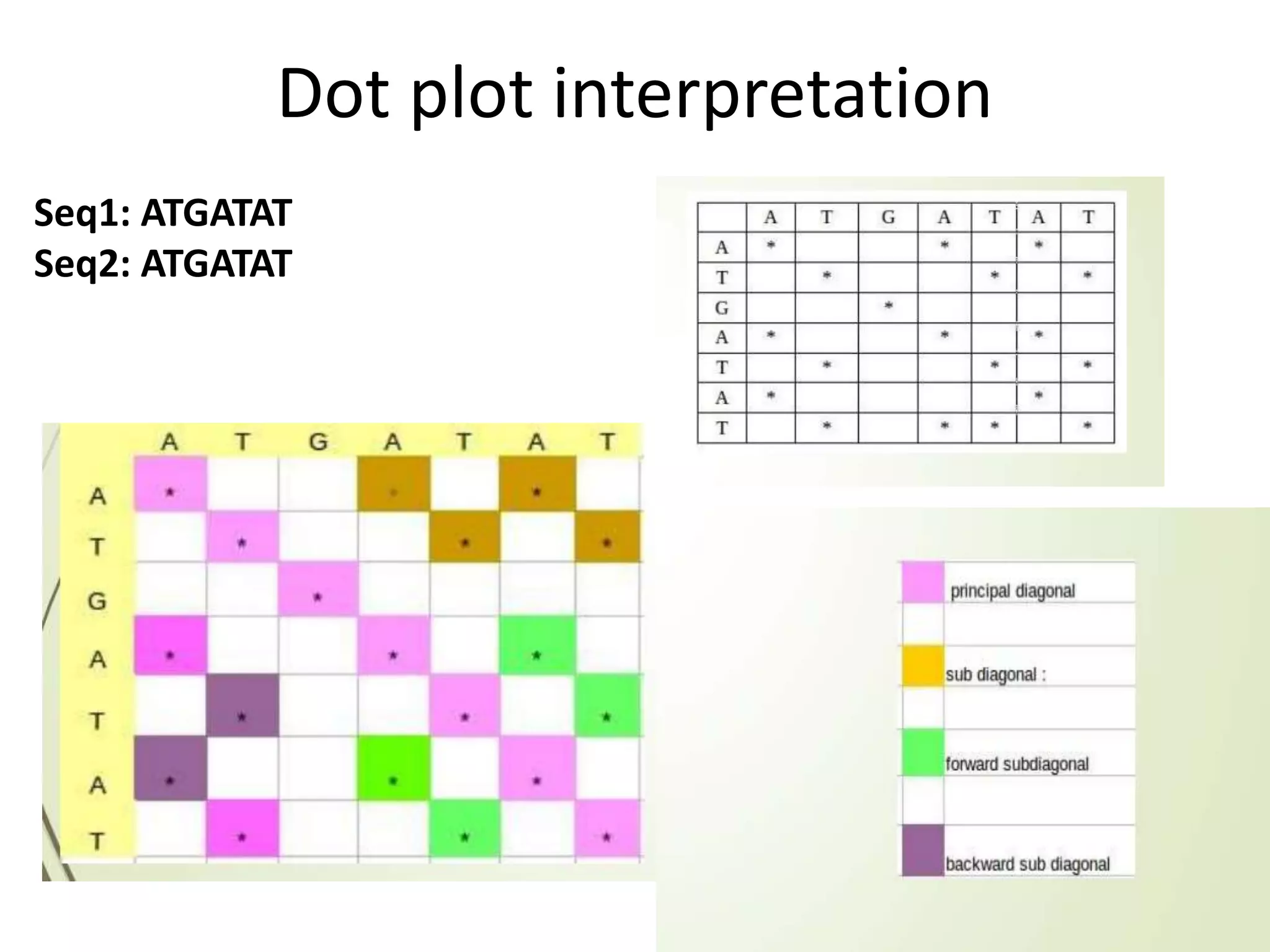
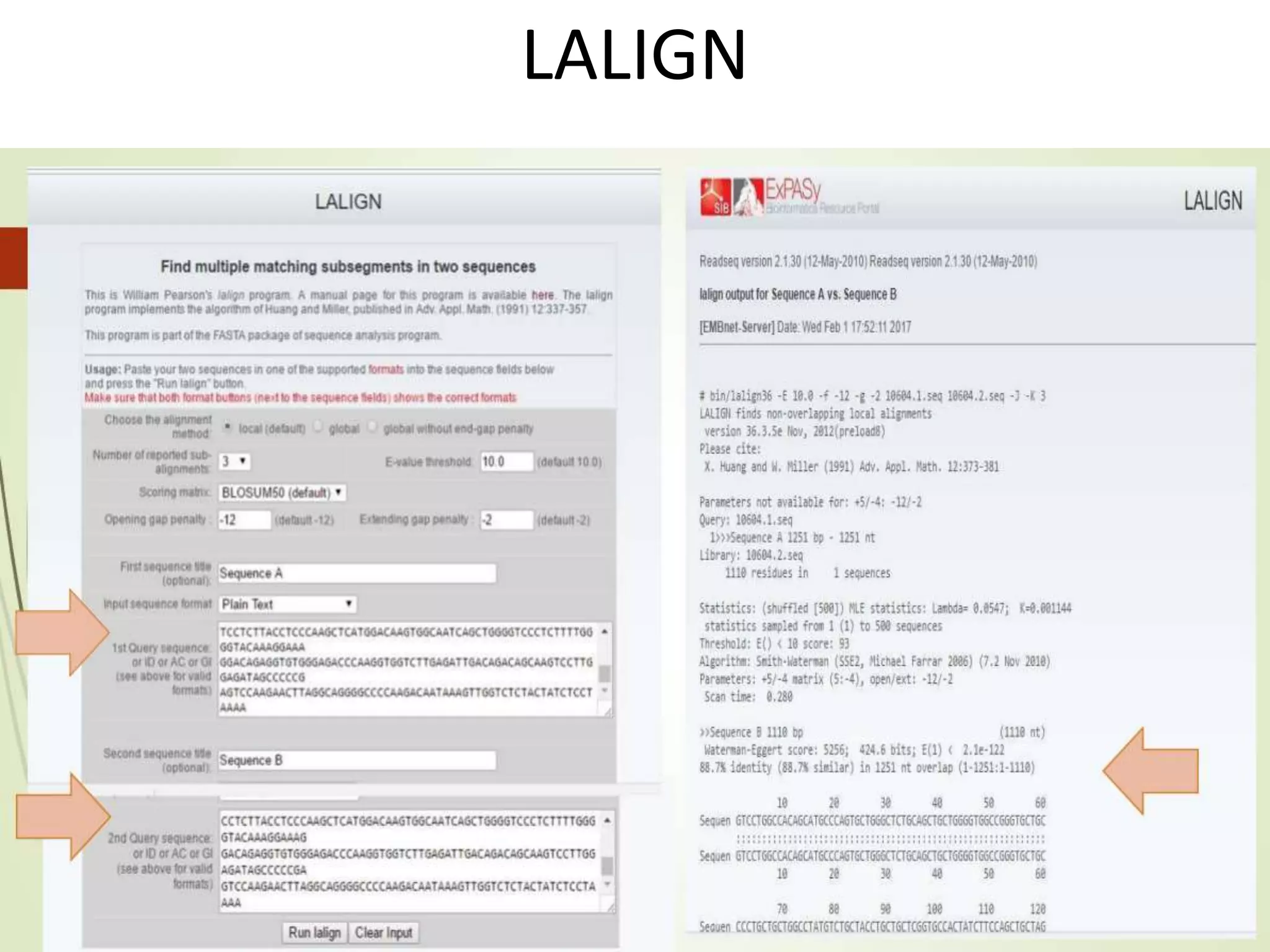
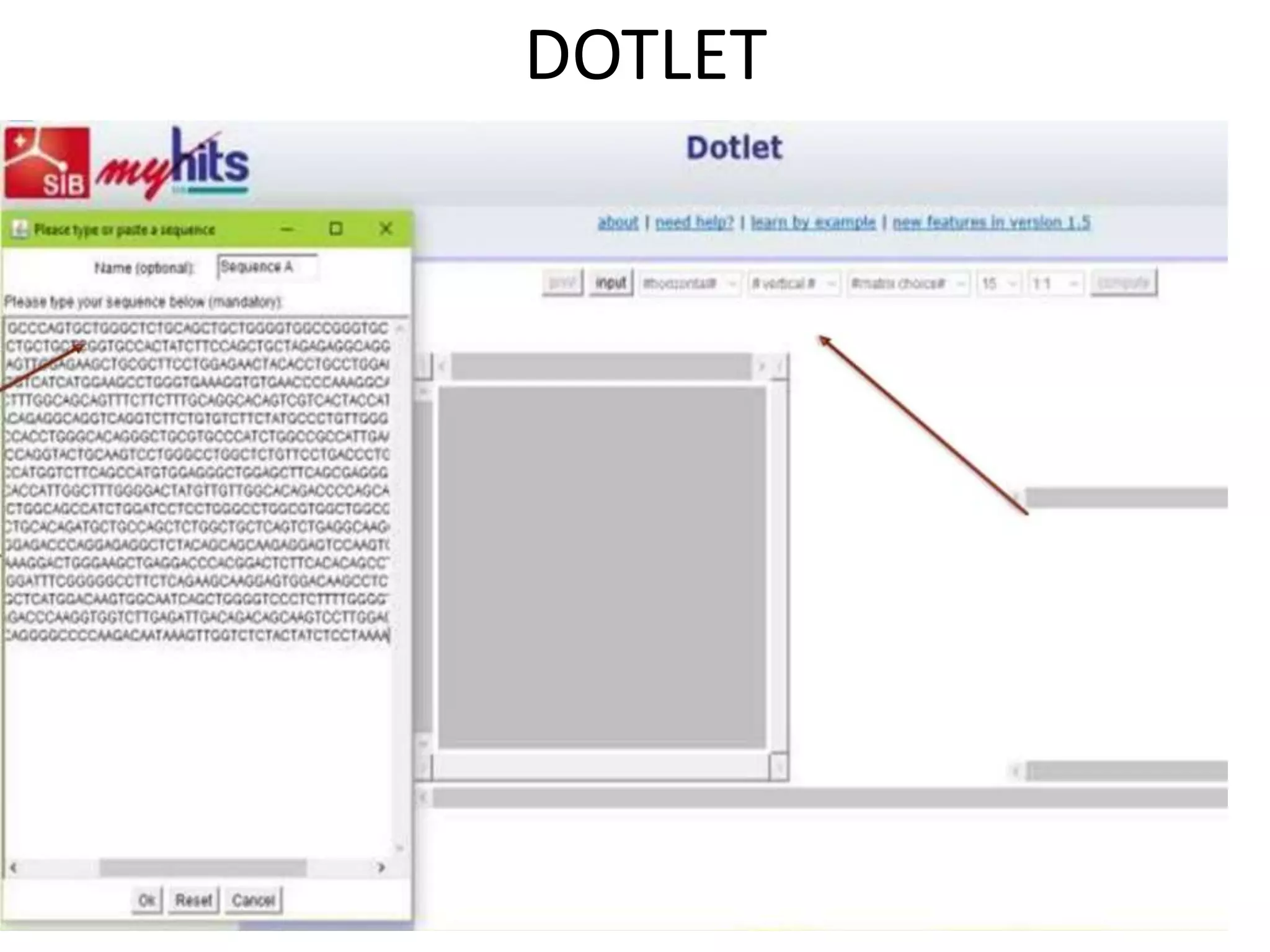
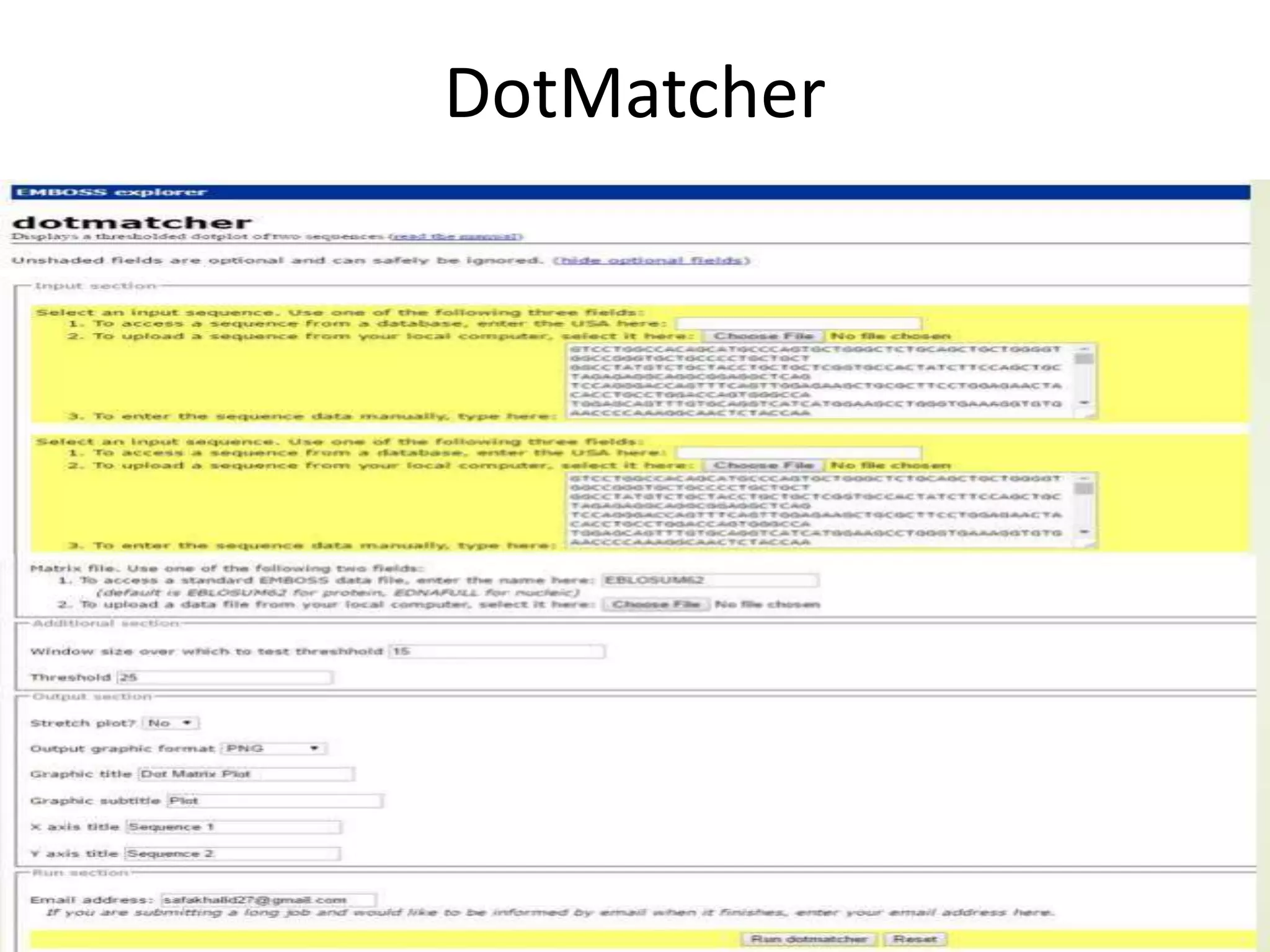
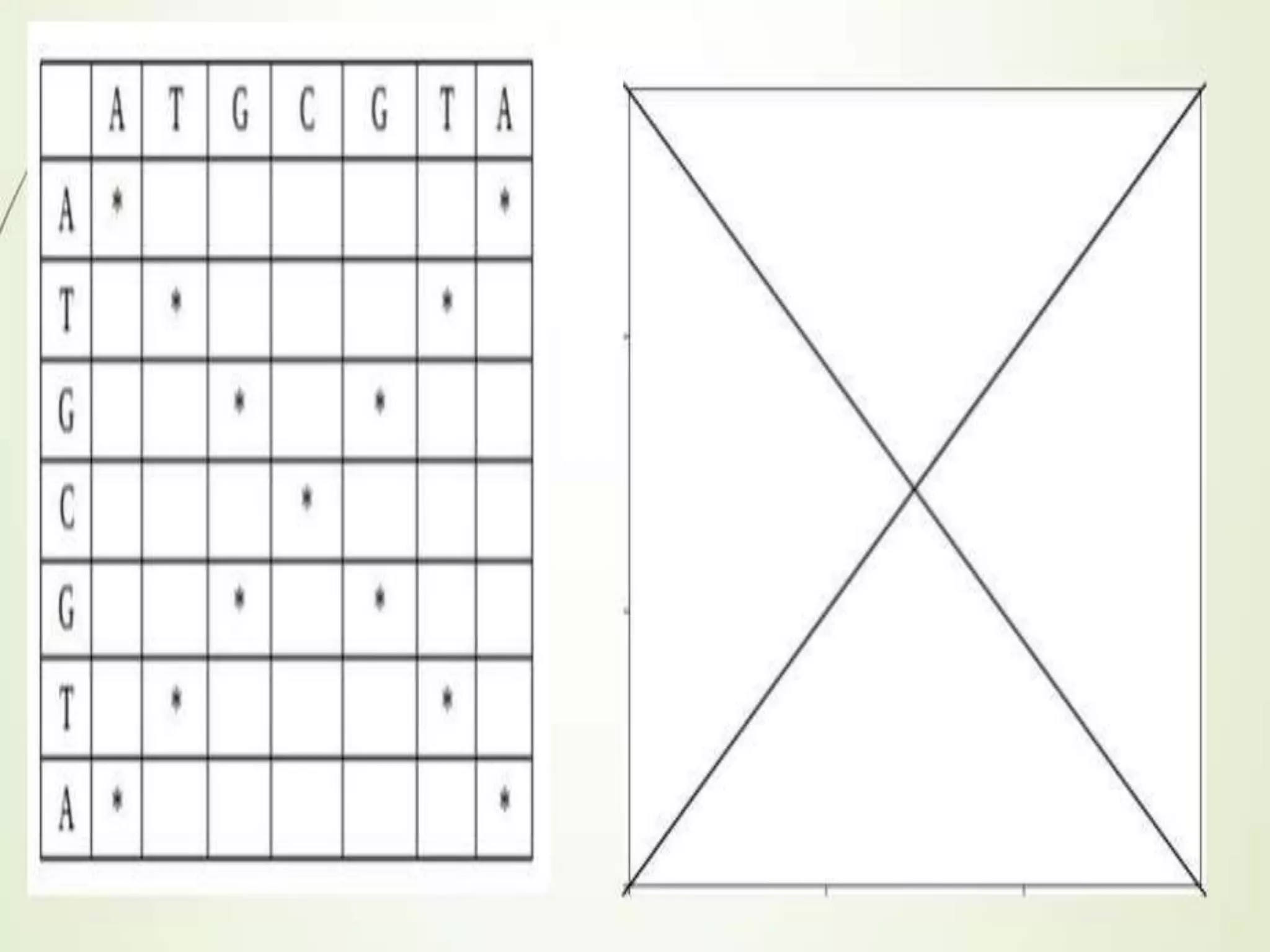
The document discusses key concepts in bioinformatics, focusing on local and global sequence alignment techniques, including the Needleman-Wunsch algorithm and local alignments for dissimilar sequences. It also covers pairwise alignment, multiple sequence alignment, phylogenetic analysis, and the use of dot plots for visualizing similarities between sequences. Additional topics include computational methods for tree building and the definitions of inverted repeats and palindromic sequences.