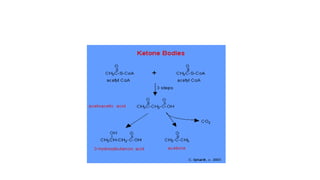
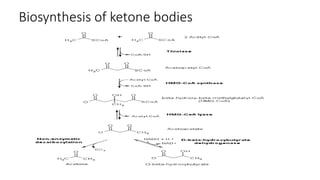
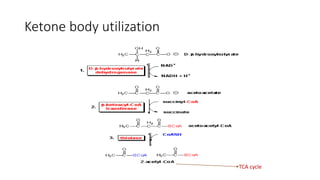
Ketone bodies are water-soluble molecules produced from the breakdown of fats in the liver during fasting. The three ketone bodies are acetoacetic acid, beta-hydroxybutyrate, and acetone. They serve as an important source of energy for tissues like heart, skeletal muscle and brain during fasting. Ketone bodies are synthesized in the liver mitochondria from acetyl-CoA and then utilized by tissues through mitochondrial enzymes. Certain genetic disorders can impair ketone body breakdown and cause health issues.




























































![Preclinical_Phase_of_Drug_Development_Lucas[1].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/preclinicalphaseofdrugdevelopmentlucas1-251107071109-7c5cab19-thumbnail.jpg?width=640&height=640&fit=bounds)




![SAMPLE BOTTLES AND SAMPLES COLLETED FOR CHEMICAL PATHOLOGY (1) [Recovered].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/samplebottlesandsamplescolletedforchemicalpathology1recovered-250503114249-98ebb71f-thumbnail.jpg?width=640&height=640&fit=bounds)




























