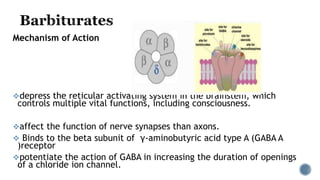
1. IV induction drugs cause rapid loss of consciousness within one arm-brain circulation time by depressing the reticular activating system when given in appropriate doses.
2. An ideal IV induction drug would have rapid onset, short duration, and few side effects. It would be water soluble, stable, and inexpensive.
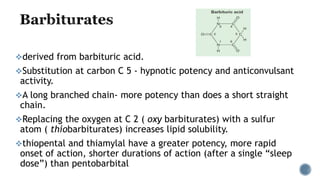
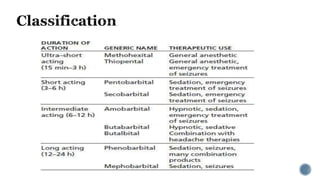
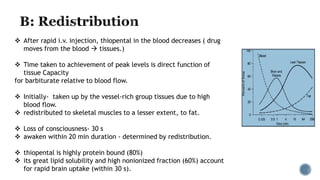
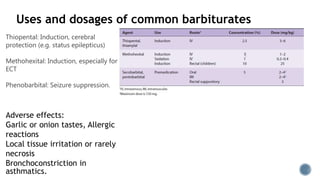
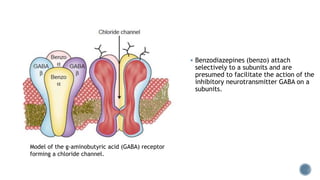
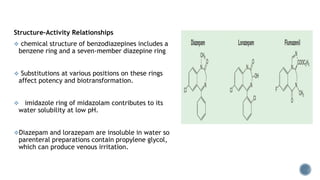
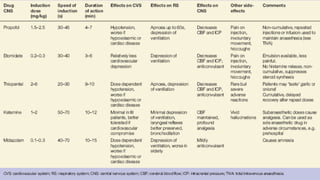
3. Common IV induction drugs include barbiturates like thiopental and methohexital, non-barbiturates like propofol and etomidate, benzodiazepines, and dissociatives like ketamine. Each drug has unique properties affecting absorption, distribution, metabolism, and excretion.