Downloaded 34 times





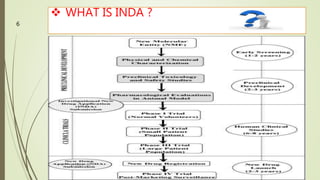
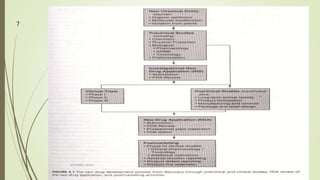

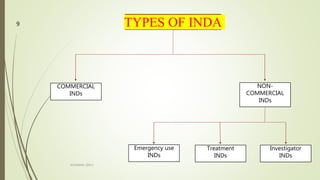
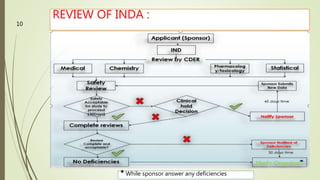













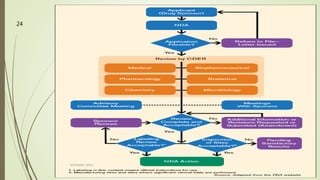









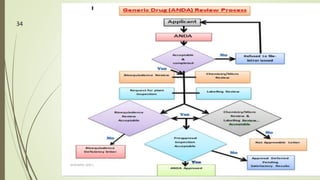
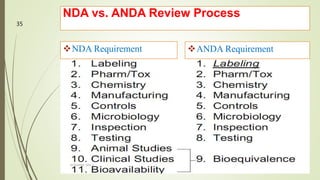










The document discusses different types of drug applications required for drug approval and marketing: Investigational New Drug Application (INDA), New Drug Application (NDA), and Abbreviated New Drug Application (ANDA). The INDA is required to begin clinical trials, the NDA contains all data from preclinical and clinical trials for new drug approval, and the ANDA allows generic versions of approved drugs to be marketed without repeating trials.

























![INVESTIGATIONAL NEW DRUG [IND] APPLICATION SUBMISSION (1).pdf](https://cdn.slidesharecdn.com/ss_thumbnails/investigationalnewdrugindapplicationsubmission1-221128141339-adad00bd-thumbnail.jpg?width=640&height=640&fit=bounds)









































