Downloaded 174 times










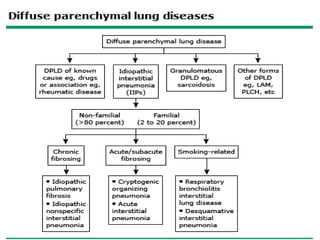


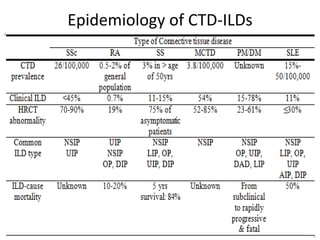
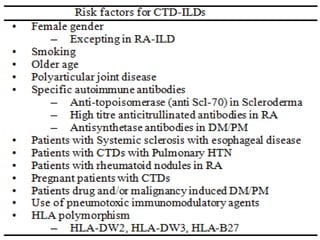




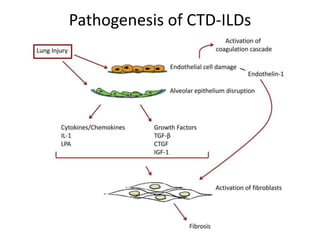





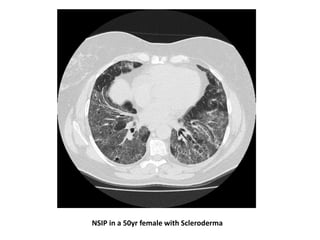



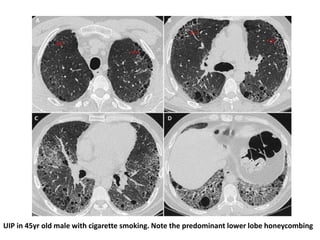



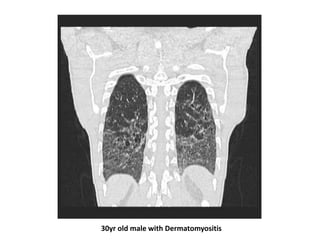



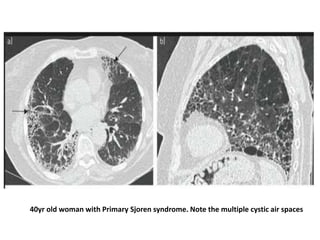



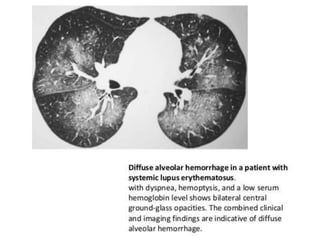

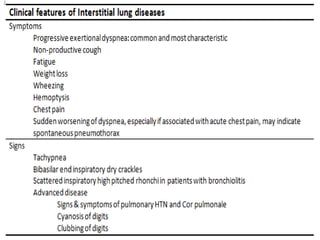
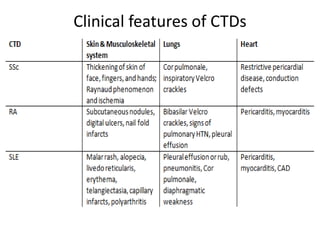
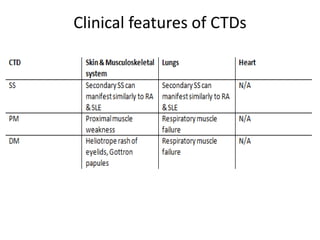
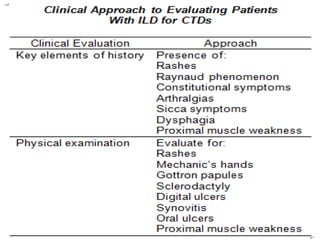
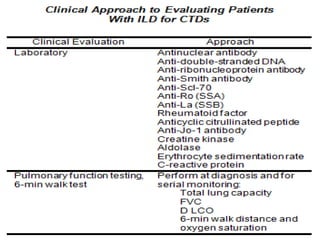

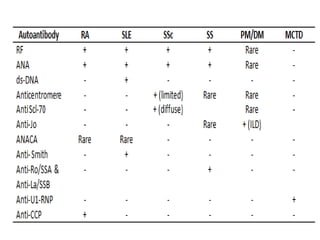









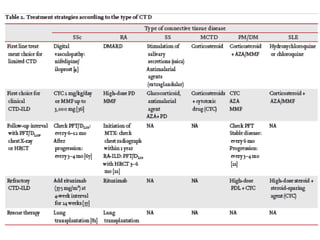
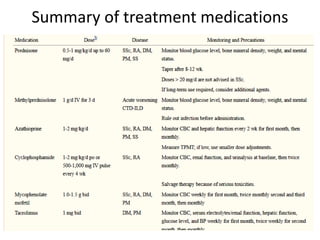







This document provides an overview of connective tissue disease (CTD)-associated interstitial lung disease (ILD). Some key points: - ILD is a common pulmonary complication in patients with CTDs like systemic sclerosis (SSc), rheumatoid arthritis (RA), and systemic lupus erythematosus (SLE). It can occur concurrently with or after diagnosis of the CTD. - The pathogenesis involves autoimmune mechanisms, genetic factors, environmental exposures, and inflammatory cytokines that cause lung inflammation and fibrosis. - SSc has the highest rate of ILD of all CTDs, affecting 40-80% of patients. Antibodies to topoisomerase I are associated with increased




































![Interstitial Lung Diseases [ILD] Approach to Management](https://cdn.slidesharecdn.com/ss_thumbnails/interstitiallungdiseases-arunvasireddy-19october2015-seminar-171016041856-thumbnail.jpg?width=640&height=640&fit=bounds)















![CASE_PRESENTATION_ON_subdural_hematoma(SDH)[1 FINAL PPT]-1.pptx](https://cdn.slidesharecdn.com/ss_thumbnails/casepresentationonsubduralhematomasdh1finalppt-1-260129172522-d405d375-thumbnail.jpg?width=640&height=640&fit=bounds)














