


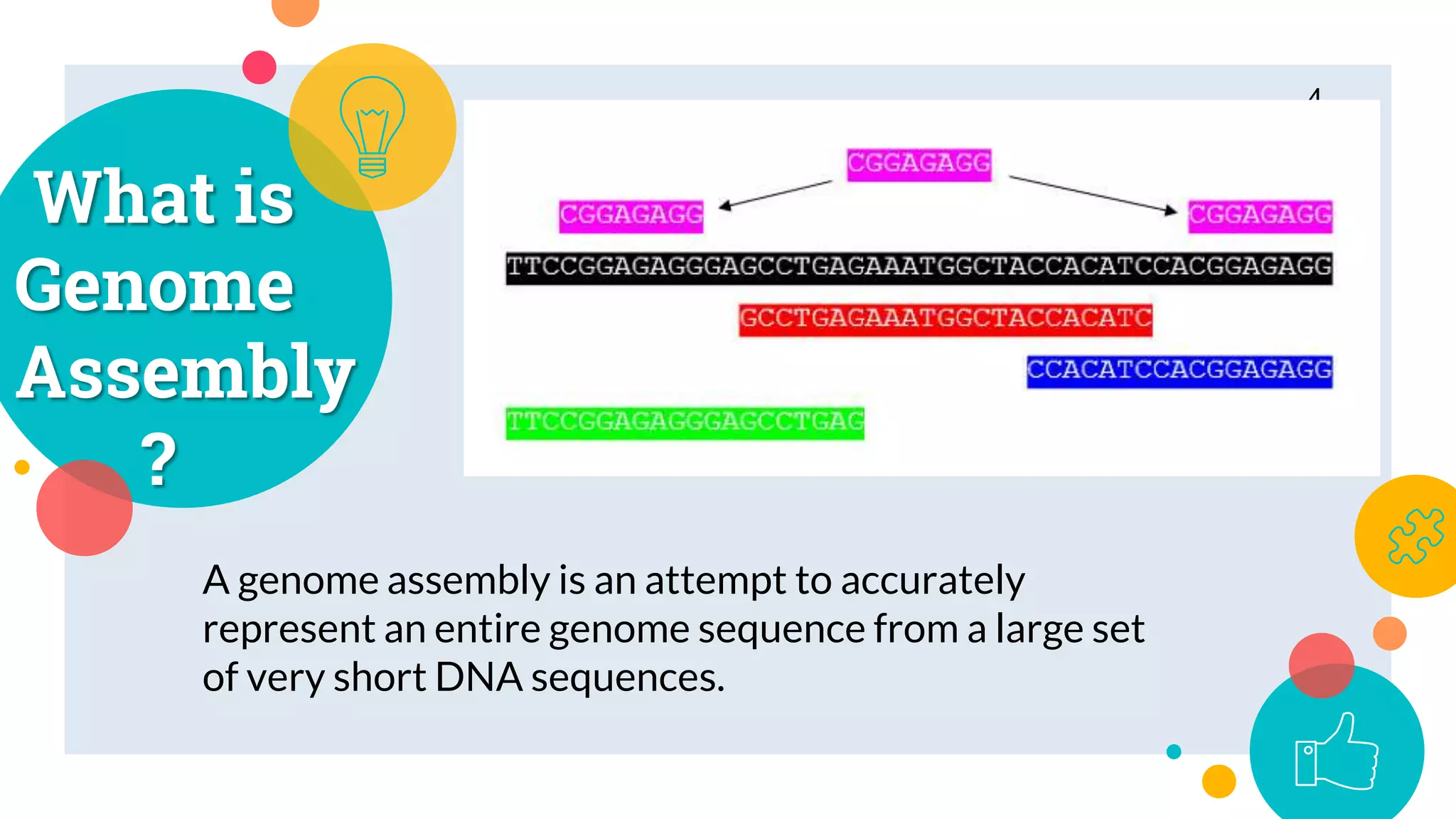

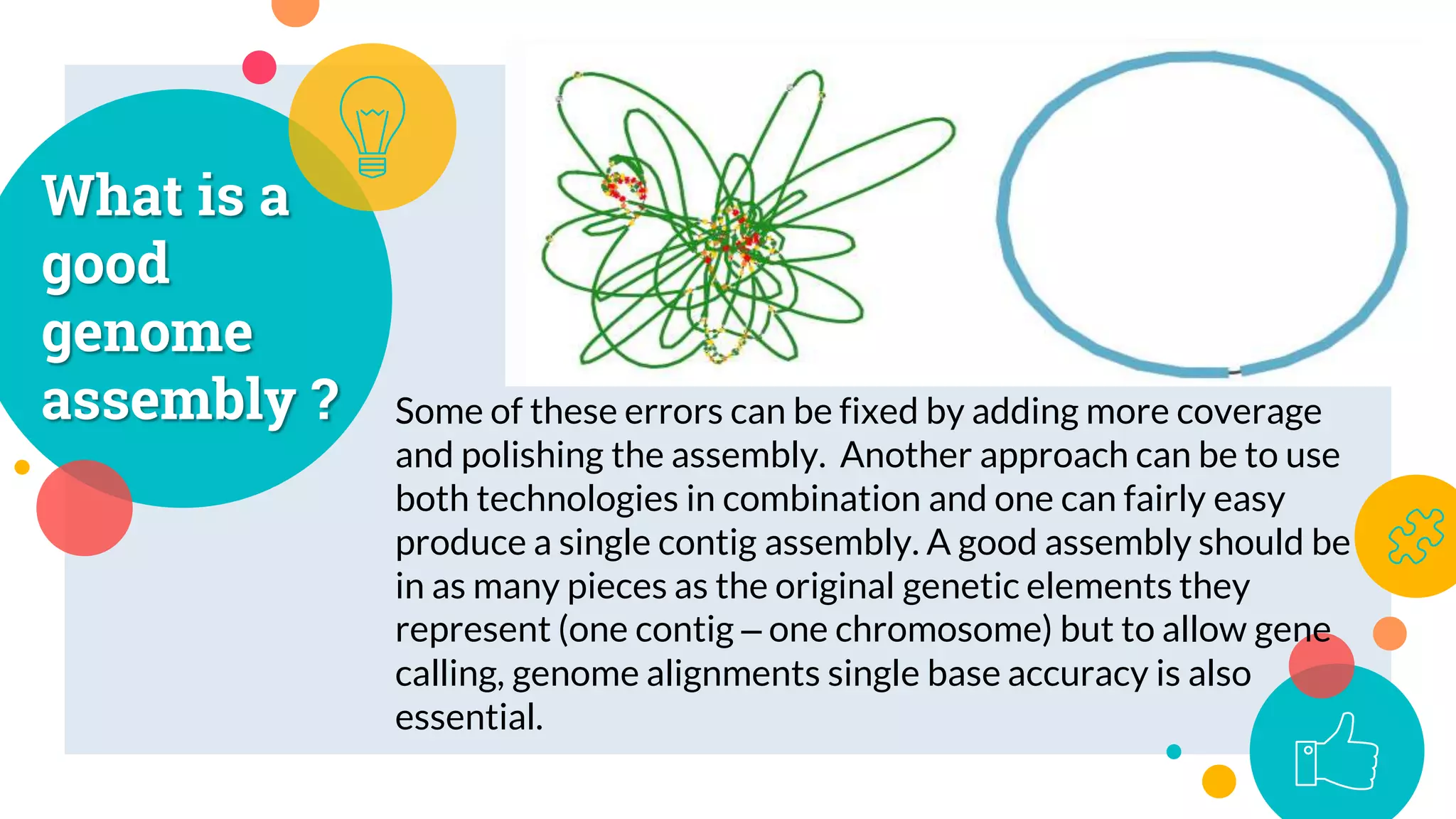


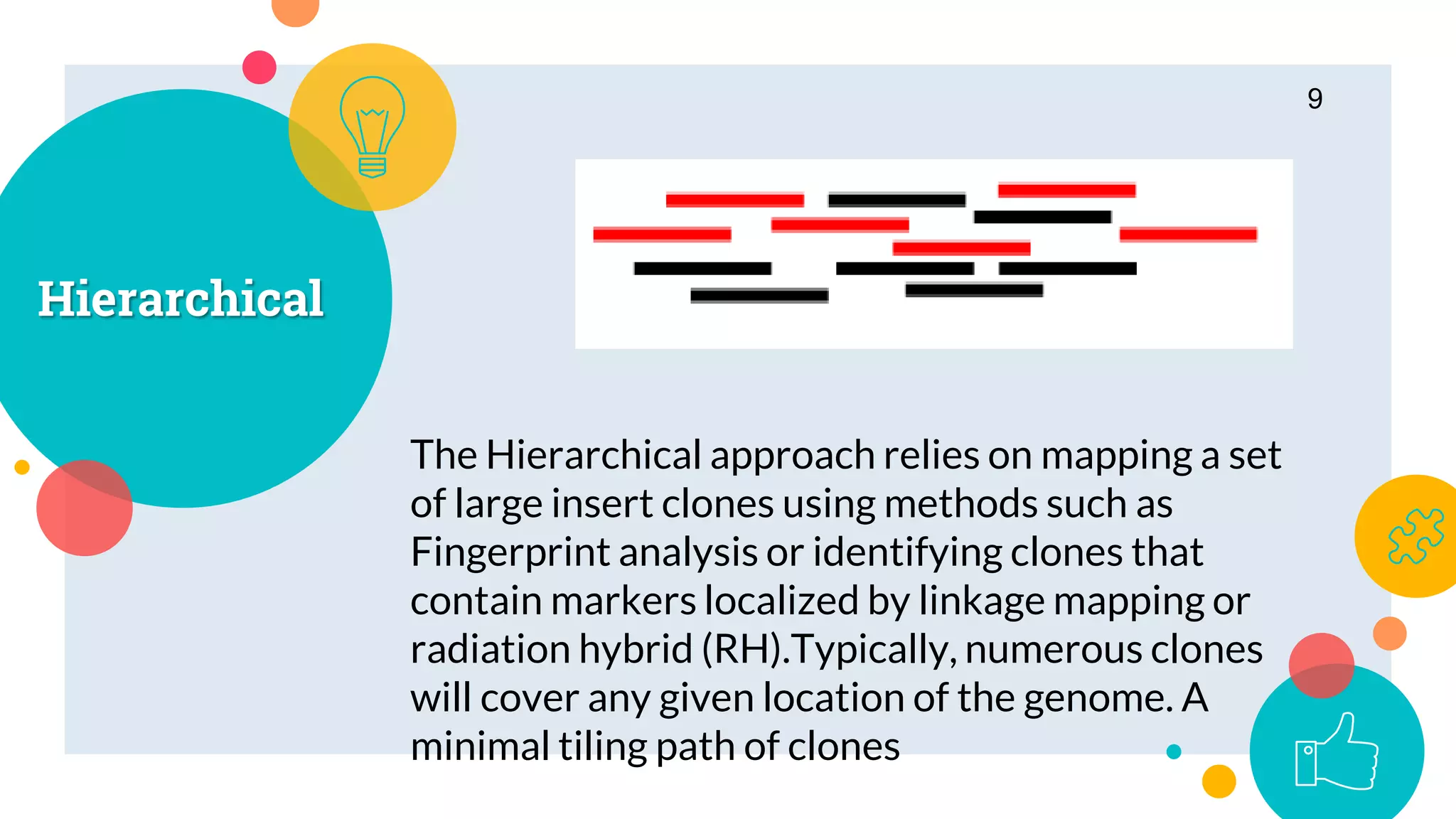
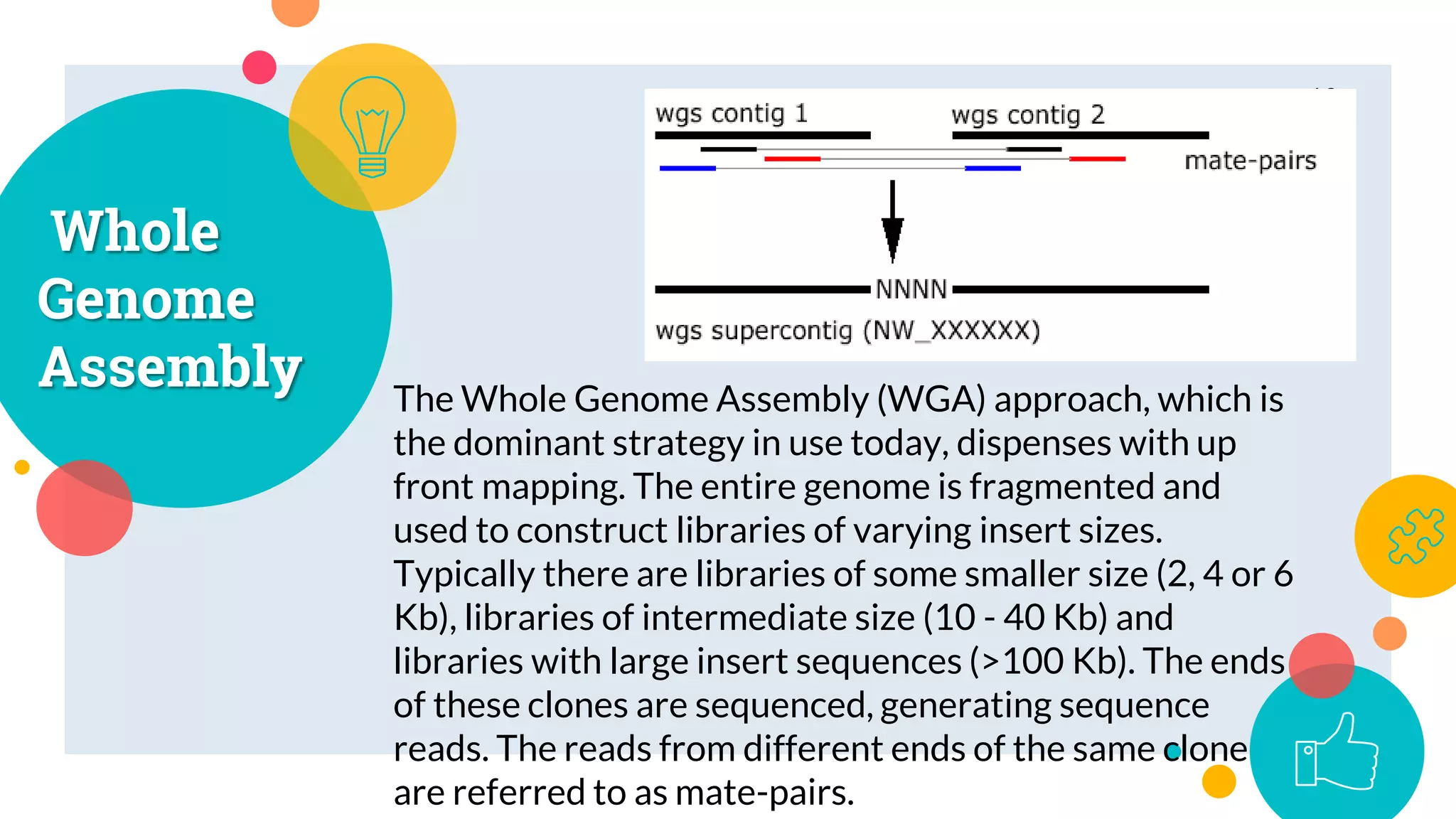
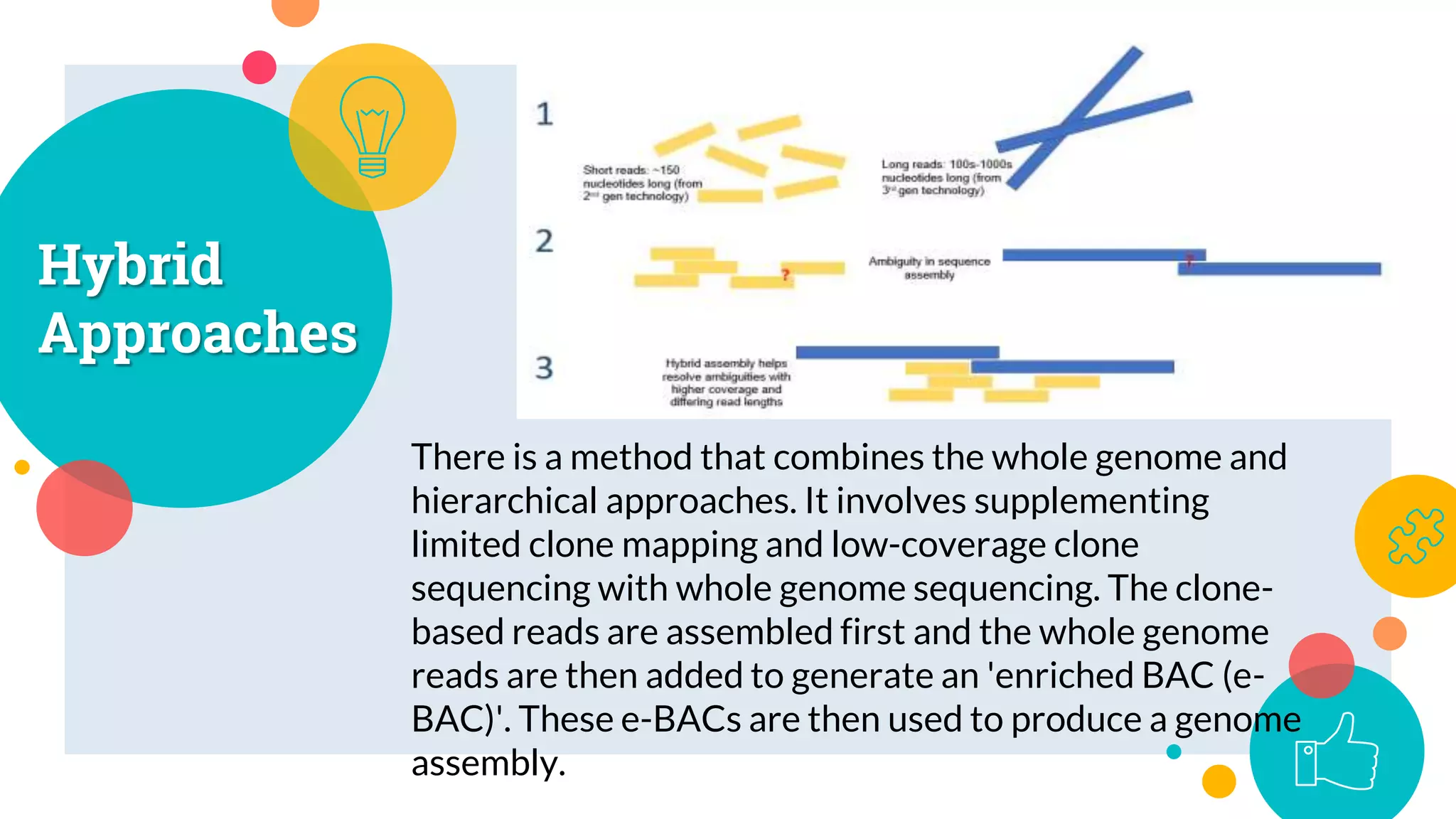
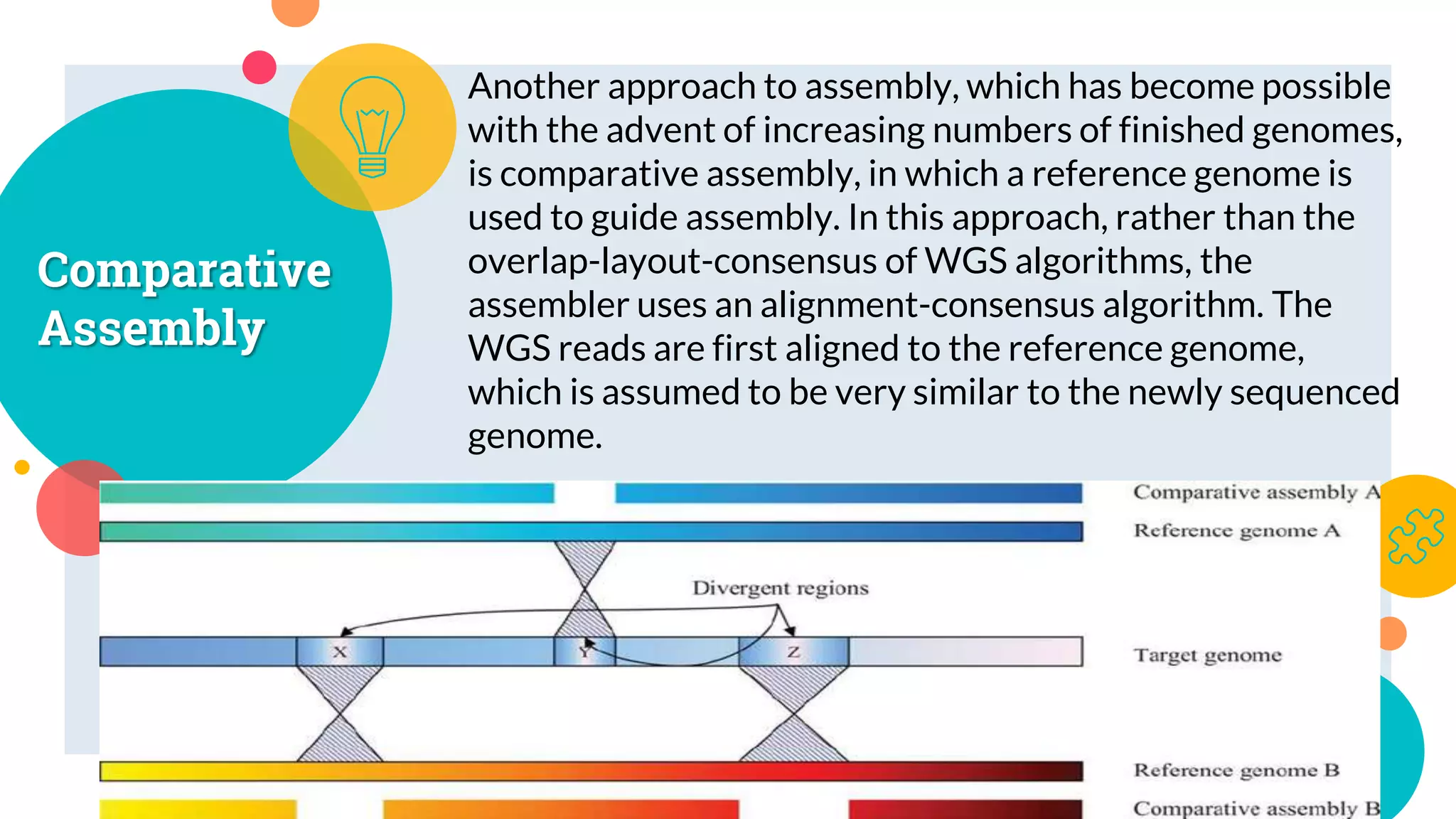



The presentation by Marjuk Ahmed Siddiki discusses genome assembly, which involves reconstructing an entire genome sequence from short DNA fragments. It covers the history and methods of genome assembly, including hierarchical, whole genome, hybrid, and comparative approaches, emphasizing the importance of accuracy and coverage in producing quality assemblies. The presentation concludes with a thanks for the attention of the audience.








































































