Hemostasis isthe process of blood clotting in areas of blood vessel
injury
Over time, a clot is lysed by the fibrinolytic system, and normal
blood flow is restored.
If clotting is impaired, hemorrhage occurs
If clotting is excessive, thrombosis occurs.
Therefore the hemostatic response needs to be rapid and regulated.
HEMOSTASIS
4.
platelet adheres tosite of vascular injury (plt provides a surface for
adhering
of clotting factors)
Activated enzyme plus substrate plus cofactor on a
reaction surface, accelerate reaction rates and provide activated
products for reaction with clotting factors further down the
coagulation cascade.
HEMOSTASIS
Most componentsof hemostasis are multifunctional;
fibrinogen
- tethers platelet to each other leading to platelet aggregation
- substrate for thrombin that forms the fibrin clot.
Platelets
- provide the reaction surface on which clotting reactions occur,
- form the plug at the site of vessel injury,
- contract to constrict and limit clot size.
7.
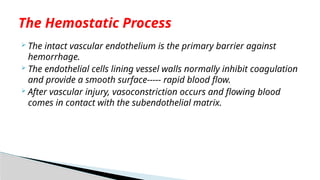
The intactvascular endothelium is the primary barrier against
hemorrhage.
The endothelial cells lining vessel walls normally inhibit coagulation
and provide a smooth surface----- rapid blood flow.
After vascular injury, vasoconstriction occurs and flowing blood
comes in contact with the subendothelial matrix.
The Hemostatic Process
8.
composed of 4major events
1- Vascular constriction
-limits the flow of blood to the area of injury.
2- Platelet aggregation
–Platelets clump when binding to exposed
collagen following rupture of the endothelial
lining of vessels.
-Platelets become activated and aggregate at the
site of injury (thrombin and fibrinogen-mediated
effects). Upon activation, platelets release ADP
and TXA2 (which activate additional platelets).
HEMOSTASIS:
9.
3-Clot formation
-to ensurestability of the initially loose platelet plug, a
fibrin mesh (also called the clot) forms and entraps the
plug.
4-Fibrinolysis
-the clot must be dissolved in order for normal blood
flow to resume following tissue repair. Clot dissolution
occurs through the action of plasmin.
HEMOSTASIS cont.
10.
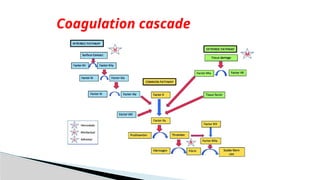
Intrinsic pathway:initiated when contact is made
between blood and exposed endothelial cell surfaces.
Extrinsic pathway: initiated upon vascular injury which
leads to exposure of tissue factor (TF) ( a sub endothelial
cell-surface glycoprotein that binds phospholipids).
Common pathway
Coagulation Phase / cascade
11.
Intrinsic Pathway
Allclotting factors are
within the blood vessels
Clotting slower
test: Activated partial
thromboplastin time
(aPTT)
Extrinsic Pathway
Initiating factor is
outside the blood
vessels - tissue
factor
Clotting - faster - in
Seconds
Test: Prothrombin
time (PT)
Coagulation Phase
12.
There existtwo systems to stop clotting:
◦ Inhibitor system
◦ Fibrinolytic system
Inhibitor system consists of:
◦ Anti-thrombin III
◦ Protein C
◦ Protein S
Protein C, S are vitamin K-dependent
What stops the clotting (anticoagulants)?
13.
Antithrombin IIIinactivates:
◦ Thrombin, Factor XIIa, XIa, Xa, IXa
During clot formation small amounts of thrombin are
swept downstream
Binds to thrombomodulin which is on the surface of the
intact endothelium
Activated protein C (APC) binds protein S,--------- inactivating
factors Va, VIIIa
Inhibitor System
14.
Plasminogen isconverted to plasmin
Plasminogen activators:
◦ t-PA
Synthesized in vascular endothelial cell
Stimulated by exercise, stasis
◦ Urokinase
Produced in kidneys
Fibrinolytic System
due todeficiency of Factor VIII (A) or Factor IX (B)
the most common severe inherited bleeding disorders.
Occurance– approximately 1 : 5,000 males,
- 85% have factor VIII deficiency
- 10–15% have factor IX deficiency
The genes for factors VIII and IX are carried on the X
chromosome
Haemophilia (A and B) (inherited bleeding disorder)
17.
Severe haemophilia
◦activity of specific clotting factor is <1%
◦ bleeding is often spontaneous (no trigger)
moderate hemophilia
◦ have factor levels of 1–5%
◦ require mild trauma to induce bleeding.
Mild hemophilia
◦ have levels >5%,
◦ require significant trauma to cause bleeding.
Classification
18.
Factors VIIIand IX participate in a complex required for the
activation of factor X.
Together with phospholipid and calcium, they form the factor X–
activating, complex
Pathophysiology
19.
After injury,the initial hemostatic event is formation of the
platelet plug, together with generation of the fibrin clot, which
prevents further hemorrhage.
In hemophilia A and B, clot formation is delayed.
Inadequate thrombin generation leads to failure to form a tightly
cross-linked fibrin clot to support the platelet plug.
Patients with hemophilia slowly form a soft, friable clot.
20.
Neither factorVIII nor factor IX crosses the placenta;
bleeding symptoms may be present from birth or may
occur in the fetus
intracranial hemorrhages,
bleeding with circumcision (males)
easy bruising,
intramuscular hematomas,
hemarthroses,(hallmark of hemophilic bleeding)
Bleeding from minor traumatic lacerations of the mouth
(torn frenulum) which may persist for hours or days
Symptoms
21.
A reducedlevel of factor VIII or factor IX
prolonged PTT.
In severe hemophilia, the PTT value is usually 2-3 times the upper
limit of normal.
platelet count, bleeding time, PT, thrombin time are normal
Laboratory findings
Recombinant factor VIII(FVIII) or recombinant
factor IX (FIX)
Dosage depends on the bleeding site
prophylaxis is the standard of care for most children with severe
hemophilia, to prevent spontaneous bleeding and early joint
deformities
Treatment
24.
chronic arthropathye.g. arthritis
development of an inhibitor to either factor VIII or
factor IX (Inhibitors are antibodies directed against factor VIII or
factor IX that block the clotting activity)
risk of transfusion-transmitted infectious diseases
complications
25.
Avoiding trauma
anticipatory guidance (use of car seats, seatbelts, and bike
helmets) and avoidance of high-risk behaviors like contact
sports
Avoid aspirin and other NSAIDs that affect platelet function.
appropriate vaccinations against hepatitis B
Preventions
26.
most commoninherited bleeding disorder,
estimated prevalence 1 : 100 - 1 : 10,000.
Patients with VWD typically present with mucosal bleeding.
A family history of either VWD or bleeding symptoms
Due to defect in or deficiency of von Willebrand factor
Von Willebrand Disease
27.
tether plateletsto injured sub-endothelium via binding sites for
platelets and for collagen
Protects FVIII from degradation in plasma
Functions of VWF
28.
mucosal bleeding,( epistaxis and menorrhagia)
easy bruising
potentially surgical bleeding
NB: diagnosis based on symptoms may be difficult, since
minor bruising and epistaxis are common in childhood
Symptoms
29.
Patients withsignificant bleeding may present with anemia, and
some patients may have thrombocytopenia.
VWF:Ag, which measures the total amount of VWF protein present
VWF activity test, typically using the ristocetin cofactor activity
assay (VWF:RCo )
Diagnosis
30.
Desmopressin (IVor Intranasal)
◦0.3 μg/kg IV
◦1 spray Intranasal (<50 kg)
Antifibrinolytics –used in Mucosal bleeding given
as PO or IV
◦ Aminocaproic acid: 100 mg/kg PO loading dose followed by
50 mg/kg every 6 hr
◦Tranexamic acid: 1,300 mg PO 3 times daily for
5 days
Treatment
31.
Local treatmentof epistaxis, such as nasal cautery or
packing, may be helpful
Iron therapy for patients with iron-deficiency anemia
Rx con’t
32.
The mostcommon cause of acute onset of thrombocytopenia in an
otherwise well child
estimated at 1 in 20,000,
1-4 wk after exposure to a common viral infection, an autoantibody
directed against the platelet surface develops with resultant sudden
onset of thrombocytopenia
recent history of viral illness.
peak age : 1-4 yr
males and females are equally affected
Peak coincides with the end of peak of viral respiratory illnesses (late
winter and early spring )
Idiopathic (Autoimmune) Thrombocytopenic Purpura (ITP)
33.
After bindingof the antibody to the platelet surface,
circulating antibody-coated platelets are recognized by the Fc
receptor on splenic macrophages, ingested, and destroyed
Most common viruses include EBV (acute ITP), HIV ( associated with
chronic ITP)
Some occurances seen with H. Pylori and following vaccination
Pathogenesis
34.
classic presentationof ITP is a previously healthy 1-4 yr
old child who has :
◦ sudden onset of generalized petechiae and purpura
There may be bleeding from the gums and mucous
membranes, with profound thrombocytopenia (platelet
count <10 × 109 /L).
h/o a preceding viral infection 1-4 wk before the onset
of thrombocytopenia
physical examination are normal, other than petechiae
and purpura
Clinical Manifestations
35.
1. No symptoms
2.Mild symptoms:
bruising and petechiae, occasional minor epistaxis, very little
interference with daily living
3. Moderate symptoms:
more severe skin and mucosal lesions, more troublesome
epistaxis and menorrhagia
4. Severe symptoms:
bleeding episodes—menorrhagia, epistaxis, melena—
requiring transfusion or hospitalization, symptoms interfering
seriously with the quality of life
Severity of ITP
36.
Severe bleedingis rare (<3% of cases)
In 70–80% of children who present with acute ITP, spontaneous
resolution occurs within 6 months.
intracranial hemorrhage (ICH) Fewer than 1%
Approx. 20% of children who present with acute ITP go on to
have chronic ITP
Prognosis
37.
Severe thrombocytopenia(platelet count <20 × 109 /L) is common, with
normal platelet size or increased, reflective of increased platelet turnover
In acute ITP the hemoglobin value, white blood cell (WBC) count, and
differential count should be normal.
(Hemoglobin may be decreased if there have been profuse nosebleeds
(epistaxis) or menorrhagia)
Bone marrow examination
◦ normal granulocytic and erythrocytic series,
◦ normal or increased numbers of megakaryocytes.
◦ Immature megakaryocytes
A direct antiglobulin test (Coombs) should be done if there is unexplained
anemia, to rule out Evans syndrome (autoimmune hemolytic anemia
and thrombocytopenia)
Diagnosis
38.
drug-dependent antibodies,
splenic sequestration
thrombocytopenia–absent radius (TAR) syndrome
hemolytic-uremic syndrome (HUS)
disseminated intravascular coagulation (DIC)
heparin-induced thrombocytopenia
Hypersplenism (in association with isolated splenomegaly )
initial manifestation of :
SLE, HIV infection,
common variable immunodeficiency,
lymphoma or autoimmune lymphoproliferative syndrome.
Ddx
39.
no datashowing that treatment affects either short- or long-term
clinical outcome of ITP
treatment appears to be capable of inducing a more rapid rise in
platelet count to the theoretically safe level of >20 × 109 /L,
although no data indicate that early therapy prevents ICH
Antiplatelet antibodies bind to transfused platelets as well as they do
to autologous platelets. Thus, platelet transfusion in ITP is usually
contraindicated unless life-threatening bleeding is present.
Treatment
40.
No therapyother than education and counseling of the family and
patient for patients with minimal, mild, and moderate symptoms
Treatment with either IVIG or corticosteroids, particularly for children
who present with mucocutaneous bleeding.
◦ A single dose of IVIG [intravenous immune globulin] (0.8-1.0
g/kg) or a short course of corticosteroids should be used as first-
line treatment.”
◦ IVIG at a dose of 0.8-1.0 g/kg/day for 1-2 days induces a rapid
rise in platelet count (usually >20 × 109 /L) in 95% of patients
41.
splenectomy in
◦Chronic ITP
◦ Life threatening hemorrhage (ICH) complicating acute ITP
rituximab (alternative to splenectomy)
42.
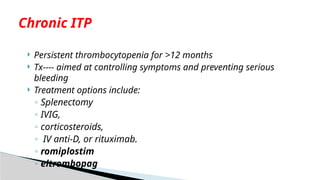
Persistent thrombocytopeniafor >12 months
Tx---- aimed at controlling symptoms and preventing serious
bleeding
Treatment options include:
◦ Splenectomy
◦ IVIG,
◦ corticosteroids,
◦ IV anti-D, or rituximab.
◦ romiplostim
◦ eltrombopag
Chronic ITP
43.
associated withimmune thrombocytopenia as the result of either an
immune process or megakaryocyte injury
Some common drugs used in pediatrics that cause thrombocytopenia
include
valproic acid
◦ phenytoin
◦ carbamazepine
◦ sulphonamides
◦ vancomycin
◦ trimethoprim-sulfamethoxazole
Drug-Induced Thrombocytopenia
44.
rare thromboticmicroangiopathy
characterized by the pentad of :
◦ fever
◦ Microangiopathic hemolytic anemia
◦ thrombocytopenia
◦ abnormal renal function
◦ CNS changes like orientation, aphasia, blindness, and seizures
Thrombotic Thrombocytopenic Purpura
45.
Laboratory findings:
microangiopathic hemolytic anemia characterized by:
◦ morphologically abnormal RBCs, with schistocytes, spherocytes, helmet cells,
elevated reticulocyte count
thrombocytopenia.
Coagulation studies are usually nondiagnostic.
Blood urea nitrogen and creatinine are sometimes elevated.
Diagnosis
is acommon cause of community-acquired acute kidney injury in
young children.
It is the most common form of thrombotic microangiopathy (TMA)
in children
Hemolytic-uremic syndrome (HUS)
48.
infection-induced, genetic,drug-induced, associated with systemic
diseases (characterized by microvascular injury)
The most common form of HUS is caused by :
Shiga toxin–producing Escherichia coli (STEC),- O157:H7 which
causes prodromal acute enteritis
commonly termed STEC-HUS or diarrhea-associated HUS.
Aetiology
49.
The reservoirof STEC is the intestinal tract of domestic animals,
usually cows.
Disease commonly is transmitted by undercooked meat or
unpasteurized (raw) milk and apple cider
contaminated municipal water supplies; petting farms; and
swimming in contaminated ponds, lakes, or pools.
With broad food distribution, Wider epidemics have been traced to
lettuce, raw spinach, and bean sprouts contaminated with STEC
50.
Genetic formsof HUS (atypical, non-diarrheal) compose the second
major category of the disease
A major feature characteristic of genetic forms of HUS is the absence of
a preceding diarrheal prodrome.
It develops slowly and unremitting once they become manifest
or they can have a relapsing pattern precipitated by an infectious illness
HUS can be superimposed on any disease associated with
microvascular injury, including malignant hypertension, SLE and
antiphospholipid syndrome.
It can also occur following bone marrow or solid organ transplantation
51.
These toxinsare easily absorbed from the colonic mucosa into the systemic
circulation,
bind to endothelial cells in the glomerulus and elsewhere, and directly cause
endothelial cell damage.
Shiga toxin can also directly activate platelets to promote their aggregation.
Mechanical injury to RBCs passing through the thrombotic microvasculature
results in a severe nonimmune anemia with a negative direct Coombs test
In each form of HUS, capillary and arteriolar endothelial injury in the kidney
leads to localized thrombosis, particularly in glomeruli, causing a direct
decrease in glomerular filtration.
Progressive platelet aggregation in the areas of microvascular injury results
in consumptive thrombocytopenia
Pathogenesis
52.
HUS (diarrheaform) is most common in preschool and school-age children,
but it can occur in adolescents and adults.
In HUS caused by toxigenic E. coli, the onset of HUS occurs 5-7 days after the
onset of gastroenteritis with fever, vomiting, abdominal pain, and diarrhea
The diarrhea is often bloody, (but not necessarily so)
Following the prodromal illness: the sudden onset of pallor, weakness, and
lethargy heralds the onset of HUS (it reflects the development of
microangiopathic hemolytic anemia )
Oliguria
patients with HUS can present with either significant dehydration or volume
overload, depending on whether the enteritis or renal insufficiency from HUS
predominates, and the amount of fluid that has been administered.
Clinical manifestation
53.
Patients withHUS who appear mildly affected at presentation can rapidly
develop severe, multisystem, life-threatening complications.
Renal insufficiency can be mild but also can rapidly evolve into severe
oliguric or anuric renal failure.( dialysis requirement in 50% of patients with
STEC-HUS
The combination of rapidly developing renal failure and severe hemolysis
can result in life-threatening hyperkalemia.
Volume overload, hypertension, and severe anemia can all develop soon
after the onset of HUS, and together can precipitate heart failure
54.
central nervous system(CNS)involvement.
significant irritability, lethargy, or non-specific encephalopathic features.
Severe CNS involvement occurs in 20% of cases.
≤
Seizures and significant encephalopathy are the most common
manifestations in those with severe CNS involvement,
Hypertension may produce an encephalopathy and seizures.
combination ofmicroangiopathic hemolytic anemia with
schistocytes, thrombocytopenia, and some degree of kidney
involvement.
Partial thromboplastin and prothrombin times are usually
normal.
The Coombs test is negative
Urinalysis typically shows microscopic hematuria and low-grade
proteinuria.
The renal insufficiency can vary from mild elevations in serum
blood urea nitrogen and creatinine to acute, anuric kidney
failure.
The presence or absence of toxigenic organisms on stool culture has little role in making the
diagnosis of diarrhea-associated STEC-HUS
Significant leucocytosis
Diagnosis
57.
Supportive care(includes careful management of fluid and
electrolytes)
prompt correction of a volume deficit
control of hypertension
early institution of dialysis if the patient becomes significantly oliguric
or anuric, particularly with hyperkalemia.
Red cell transfusions are usually required because hemolysis can be
brisk and recurrent until the active phase of the disease has resolved.
Antibiotic therapy to clear enteric toxigenic organisms (STEC) not
recommended----- can result in increased toxin release, potentially
exacerbating the disease.
Treatment
58.
Most patientswith diarrhea-associated HUS recover completely,
with little risk of long-term sequelae.
Patients with hypertension, any level of renal insufficiency, or
residual urinary abnormalities persisting a year after an episode
of diarrhea-positive HUS (particularly significant proteinuria)
require careful follow-up.
Patients who have recovered completely with no residual urinary abnormalities after 1
yr are unlikely to manifest long-term sequelae
59.
most commonvasculitis of childhood
also referred to as IgA vasculitis , based on the presence of
vasculitis with predominance of IgA deposits affecting small
vessels
incidence : estimated at 14-20 per 100,000 children per year
affects males more than females.
Approximately 90% of HSP cases occur in children, between 3 -
10 yrs
Many cases of HSP follow a documented upper respiratory
infection.
Henoch-Schönlein Purpura (HSP)
60.
The exactpathogenesis of HSP remains unknown
preceding upper respiratory infectious triggers such as group A
β-hemolytic streptococcus, Staphylococcus aureus, mycoplasma,
and adenovirus have been suspected
The common finding of deposition of IgA, suggests that HSP is a
disease mediated by IgA and IgA immune complexes
genetic component. has been linked to HSP nephritis
61.
The hallmarkof HSP is its rash :
palpable purpura starting as pink macules or wheals and
developing into petechiae, raised purpura, or larger
ecchymoses.
The skin lesions are usually symmetric and occur in gravity-
dependent areas (lower extremities), extensor aspect of the
upper extremities or on pressure points (buttocks) .
The skin lesions often evolve in groups, typically lasting 3-10
days, and may recur up to 4 mo after initial presentation.
Clinical features
62.
Subcutaneous edemalocalized to the dorsa of hands and feet,
periorbital area, lips, scrotum, or scalp
Musculoskeletal involvement, including arthritis and arthralgias, is
common with predilection for large joints such as the knees and
ankles, and does not lead to deformities
Periarticular swelling and tenderness without erythema or
effusions are common. The arthritis usually resolves within 2 wk
but can recur.
63.
Gastrointestinal (GI)manifestations include :
- abdominal pain,
- vomiting,
- diarrhea,
- paralytic ileus,
- and melena.
- Intussusception, mesenteric ischemia, and intestinal perforation are
rare but serious complications.
Renal involvement:
manifesting as microscopic haematuria, proteinuria, hypertension,
frank nephritis, nephrotic syndrome, and acute or chronic renal failure.
64.
Neurologic manifestationsof HSP, caused by hypertension or
central nervous system (CNS) vasculitis:
- intracerebral hemorrhage,
- seizures,
- headaches,
- depressed level of consciousness,
- cranial or peripheral neuropathies,
- and behavior changes.
Other, less common potential manifestations of HSP are
inflammatory eye disease, carditis, pulmonary hemorrhage,
orchitis, and testicular torsion.
65.
European League AgainstRheumatism/Pediatric
Rheumatology European Society Criteria
Palpable purpura and 1or more of the following criteria must be
present:
◦ Abdominal pain (acute, diffuse, colicky pain)
◦ Arthritis or arthralgia
◦ Biopsy of affected tissue demonstrating predominant IgA deposition
◦ Renal involvement (proteinuria >3 g/24 hr), haematuria or red cell casts
Diagnosis
No laboratory findingis diagnostic of HSP
leukocytosis,
thrombocytosis,
mild anemia,
elevations of erythrocyte sedimentation rate (ESR) and C-reactive protein
(CRP).
The platelet count is normal in HSP .
Occult blood is frequently found in stool specimens
Ultrasound is often used in the setting of GI complaints to look for bowel
wall edema or the rare occurrence of an associated intussusception.
Barium enema can also be used to both diagnose and treat
intussusception
Laboratory Findings
68.
Treatment formild and self-limited HSP is supportive,
- ensuring adequate hydration,
- nutrition,
- analgesia.
Corticosteroids --- significant GI involvement or other life-
threatening manifestations.
oral prednisone (1-2 mg/kg/day),
or in severe cases, intravenous (IV) methylprednisolone for 1-2 wk,
It reduces abdominal and joint pain but do not alter overall prognosis.
chronic HSP renal disease is managed with a variety of
immunosuppressants, (ie azathioprine, cyclophosphamide,
cyclosporine, and mycophenolate mofetil)
Treatment
69.
intussusception andintestinal perforation
Renal disease is the major long-term complication
Complications
70.
result inconsumption of clotting factors, platelets, and
anticoagulant proteins.
Consequences :
- widespread intravascular deposition of fibrin, leading to tissue
ischemia and necrosis, a generalized hemorrhagic state, and
microangiopathic hemolytic anemia.
Disseminated intravascular coagulopathy (DIC)
71.
Activation andrelease of cytokines and chemokines alter endothelial function to
a more prothrombotic state
enhancing the formation of microvascular thromboses, with resultant
consumption of pro- and anticoagulant proteins.
Excessive activation of clotting consumes both the physiologic anticoagulants
(protein C, protein S, and AT III) and the procoagulants
resulting in a deficiency of factor V, factor VIII, prothrombin, fibrinogen, and
platelets.
Typically, the clinical result of this sequence of events is hemorrhage
Pathogenesis
72.
Causes
INFECTIONS New born
Meningococcemia (purpura
fulminans)
Bacterial sepsis (staphylococcal,
streptococcal, Escherichia coli ,
Salmonella )
Rickettsia (Rocky Mountain spotted
fever)
Viruses (cytomegalovirus, herpes
simplex, hemorrhagic fevers)
Malaria
Fungi
Maternal toxemia
Bacterial or viral sepsis (group B
streptococcus, herpes simplex
virus)
Abruptio placentae
Severe respiratory distress
syndrome
Necrotizing enterocolitis
Erythroblastosis fetalis
Fetal demise of a twin
73.
TISSUE INJURY GASTROINTESTINALDISORDERS
Central nervous system trauma
(massive head injury)
Multiple fractures with fat emboli
Crush injury
Profound shock or asphyxia
Hypothermia or hyperthermia
Massive burns
Fulminant hepatitis
Ischemic bowel
Pancreatitis
74.
shock .
Bleedingfrequently first occurs from sites of venipuncture or
surgical incision.
The skin may show petechiae and ecchymoses.
Tissue necrosis may involve many organs and can be most
spectacularly seen as infarction of large areas of skin,
subcutaneous tissue, or kidneys.
Anemia caused by hemolysis may develop rapidly because of
microangiopathic hemolytic anemia.
Clinical manifestation
75.
Certain coagulation factors(factorsII, V, and VIII; fibrinogen)
and platelets consumed
prolongation of the prothrombin (PT)
Prolonged partial thromboplastin (PTT)
Prolonged thrombin (TT) times.
Platelet counts may be profoundly depressed.
The blood smear may contain fragmented, burr and helmet-
shaped red blood cells (schistocytes).
fibrinogen degradation products (D-dimers)appear in the blood.
Laboratory
76.
treat thetrigger that caused DIC
restore normal homeostasis by correcting the shock,
acidosis, and hypoxia that usually complicate DIC.
Blood components in patients with hemorrhage
consist of:
◦ platelet infusions (for thrombocytopenia),
◦ cryoprecipitate (for hypofibrinogenemia),
◦ fresh-frozen plasma (for replacement of other coagulation factors
and natural inhibitors)
Treatment
#5 Active clotting is controlled by negative feedback loops that inhibit the clotting process when the procoagulant process comes in contact with intact endothelium.
#14 t-PA tissue plasminogen activator
inhibitors of the inhibitor system exists so as body can clot when needed.
#15 Known as extrinsic pathway because the pathway of blood coagulation activated by tissue factor which is a protein extrinsic to the blood.
#19 When untreated bleeding occurs in a closed space, such as a joint, cessation of bleeding may be the result of tamponade .
With open wounds, in which tamponade cannot occur, profuse bleeding may result in significant blood loss.
#27 VWF that facilitates its ability to bind platelets through a binding site on platelet glycoprotein Ib (GPIb).
This enables VWF to recruit platelets to the site of clot formation
#34 The parents often state that the child was fine yesterday and now is covered with bruises and purple dots
#35 When the onset is insidious, especially in an adolescent, chronic ITP or the possibility of a systemic illness, such as systemic lupus erythematosus (SLE), is more likely
#50 The latter feature likely explains the association of many infectious agents with HUS
#51 Microangiopathic hemolytic anemia results from mechanical damage to red blood cells as they pass through the damaged and thrombotic microvasculature
#63 However, progression to end-stage renal disease (ESRD) is uncommon in children (1–2%)
#68 ESRD develops in <5% of children with HSP nephritis
#72 Any life-threatening severe systemic disease associated with hypoxia, acidosis, tissue necrosis, shock, or endothelial damage may trigger DIC.
#75 The D-dimer is formed by fibrinolysis of a cross-linked fibrin clot. The D-dimer assay is as sensitive as the fibrinogen degradation product test and more specific for activation of coagulation and fibrinolysis.






































![ No therapy other than education and counseling of the family and
patient for patients with minimal, mild, and moderate symptoms
Treatment with either IVIG or corticosteroids, particularly for children
who present with mucocutaneous bleeding.
◦ A single dose of IVIG [intravenous immune globulin] (0.8-1.0
g/kg) or a short course of corticosteroids should be used as first-
line treatment.”
◦ IVIG at a dose of 0.8-1.0 g/kg/day for 1-2 days induces a rapid
rise in platelet count (usually >20 × 109 /L) in 95% of patients](https://image.slidesharecdn.com/bleedingdisordersedited-251123070431-a9a3304a/85/Bleeding-disorders-in-internal-medicine-pro-40-320.jpg)
























































































![CTEV [ clubfoot] DR ARUN LAL ,DR MOHAMED ASHRAF travancore medical college k...](https://cdn.slidesharecdn.com/ss_thumbnails/ctevclubfootdrarunlaldrmohamedashraftravancoremedicalcollegekollamkeralaindia-260208063247-18fc466c-thumbnail.jpg?width=640&height=640&fit=bounds)




